阿托伐他汀Word文档格式.docx
《阿托伐他汀Word文档格式.docx》由会员分享,可在线阅读,更多相关《阿托伐他汀Word文档格式.docx(12页珍藏版)》请在冰豆网上搜索。
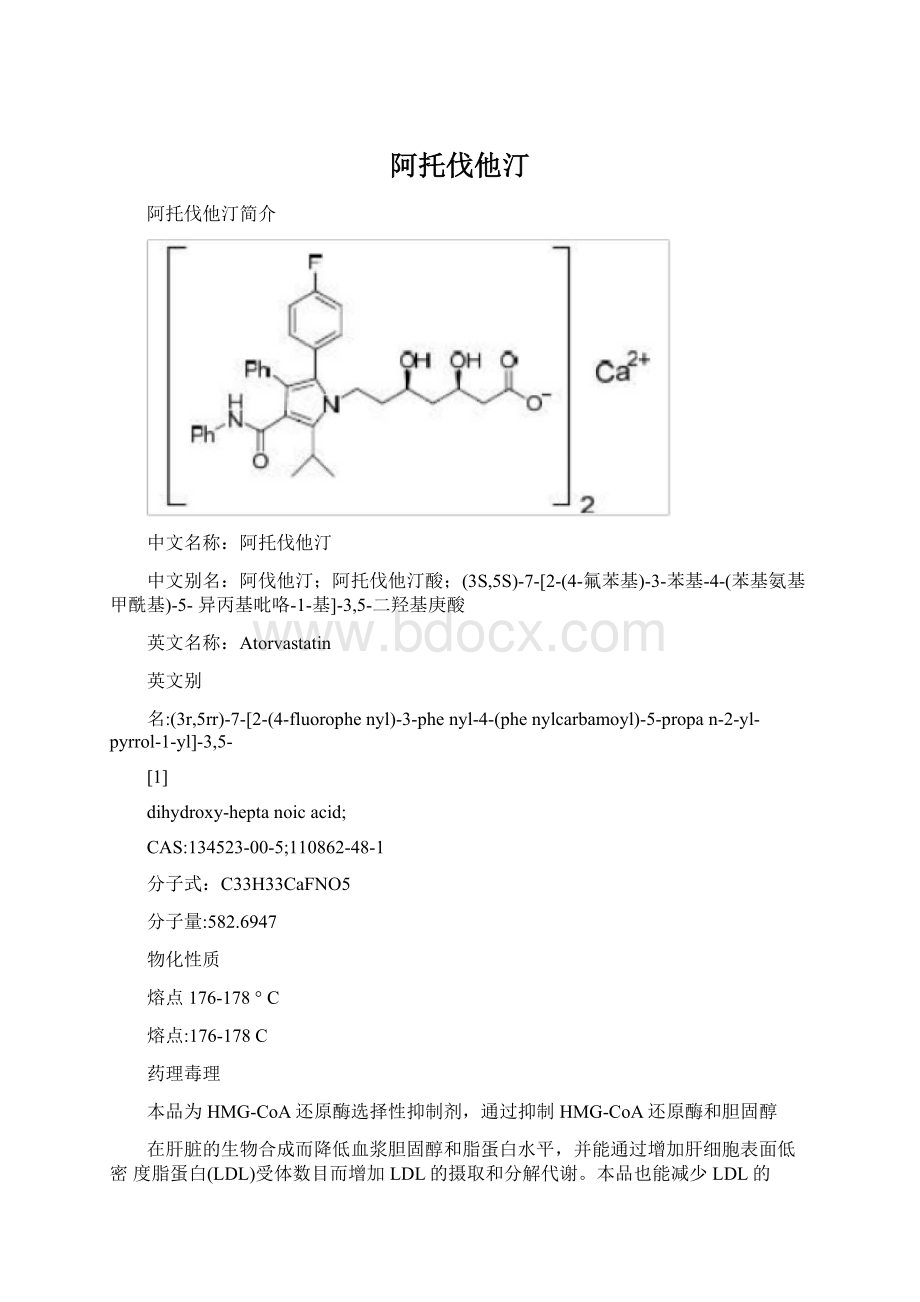
C
熔点:
176-178C
药理毒理
本品为HMG-CoA还原酶选择性抑制剂,通过抑制HMG-CoA还原酶和胆固醇
在肝脏的生物合成而降低血浆胆固醇和脂蛋白水平,并能通过增加肝细胞表面低密度脂蛋白(LDL)受体数目而增加LDL的摄取和分解代谢。
本品也能减少LDL的
生成和其颗粒数。
本品还能降低某些纯合子型家族性高胆固醇血症(FH)的低密度脂蛋白胆固醇(LDL-C)水平,而一类型的人群对其他类型的降脂药物治疗很少有应答。
本品能降低纯合子和杂合子家族性高胆固醇血症、非家族性高胆固醇血症以及混合性脂类代谢障碍患者的血浆总胆固醇(TC)、LDL-C和载脂蛋白B(ApoB),还能降低极低密度脂蛋白胆固醇(VLDL-C)和三酰甘油(TG)的水平,并能不同程度地提高血浆高密度脂蛋白胆固醇(HDL-C)和载脂蛋白A1(ApoA1)的水平。
1.9药代特征
吸收:
口服后迅速吸收,1-2小时内达到最大血浆浓度,吸收程度随口服剂量的增加而成正比例地增加。
绝对生物利用度约为12%抑制HMG-Co还原酶的全
身利用度约为30%无论是否与食物同时服用或在一天中无论何时服用,其降低血浆LDL-C的效果都相似。
分布:
平均分布容积是381升,其中98%以上与血浆蛋白结合。
代谢:
阿托伐他汀在体内被代谢成为邻羟基化和对羟基化代谢产物,以及各
种B-氧化
产物:
其对循环HMG-Co还原酶抑制活性大约70%源于活性代谢产物。
消除:
阿托伐他汀及其代谢产物通过肝脏和/或肝外途径代谢后主要经胆汁排除。
其平均血浆清除半衰期为14小时,因活性代谢产物的作用,其对HMG-Co还原酶抑制活性的半衰期达20-30小时。
适应证
杂合子家族性或非家族性高胆固醇血症和混合性高脂血症,也用于纯合子高胆固醇血症。
不良反应
本品可被较好地耐受,不良反应多为轻度和一过性,最常见的是便秘、腹胀、消化不良和腹痛⑴。
因本品的不良反应而停药者<
2%。
其他有ALT升高(0.7%),发生在用药16周内。
相互作用
本品与含有炔诺酮和炔雌醇的口服避孕药合用时,能分别使诺酮和炔雌醇的
AUC增加30%和20%;
与红霉素合用时,本品的血浆浓度约增高40%;
与地高辛合用时,多次给药后,地高辛的稳态血药浓度增加约20%,应对地高辛浓度进行监测,与考来烯胺(消胆胺)合用时,本品的血浆浓度降低约25%,但降低LDL-C的效果较单用本品或考来替泊的效果都大。
本品与环抱菌素、烟酸、红霉素及吡咯类抗真菌药合用,发生肌病的危险性增加。
出血性卒中风险:
一项称为SPARCL(StrokePreventionbyAggressiveReductioninCholesterolLevels)研究的析因分析显示,近期出现过出血性卒中或腔隙性脑梗死(无冠心病)的患者,每日服用80mg阿托伐他汀可能增加出血性卒中风险。
在SPARCL研究中,对4731名卒中或短暂性脑缺血发作患者前6个月内每日服用阿托伐他汀80mg片剂或安慰剂的疗效进行了评估。
这些患者均无冠心病史。
根据卒中亚型的析因分析,与安慰剂相比,阿托伐他汀80mg降低了缺血性卒中的发
生几率(218/2365,9.2%vs.274/2366,11.6%,p=0.01),但增加了出血性卒中的发生几率(55/2365,2.3%vs.33/2366,1.4%,p=0.02)。
在曾出现过出血性卒中或腔隙性脑梗死的患者中,出血性卒中风险的增加尤为显著。
对于曾经出现过出血性卒中或腔隙性脑梗死的患者,服用阿托伐他汀80mg
的风险/收益尚未确定,因此,在开始使用阿托伐他汀治疗前应认真考虑出血性卒中的潜在风险。
用法用量
可在任何时间单剂量服用,进食或非进食时均可。
起始剂量为10mg,qd,剂量
范围为10〜80mg•d-1。
根据治疗目标和治疗反应采取个体化治疗方案。
对肾功能不全患者,不必调整剂量。
注意事项
活动性肝病或原因不明的转氨酶持续升高患者及对本品的任何成分过敏者、孕
妇和哺乳期妇女禁用。
①使用本品治疗前、治疗6周及12周或增加药物剂量后进行肝功能检测,并在以后定期测定肝功能。
②治疗过程中出现弥漫性肌痛、肌肉触痛或无力,特别是伴有全身不适或发热时,或/和肌酸磷酸激酶(CPK)水平明显升高,应考虑肌病的可能性。
如为肌病,应停止本品的治疗。
③使用本品前,应通过适当饮食、运动和减轻肥胖的体重等方法控制高胆固醇血症,并治疗其他原发疾病。
④本品过量无特殊
治疗。
一旦发生过量,应予以对症及相应的支持疗法,血液透极不能显著增加其清除。
⑤开始治疗和/或增加剂量后2〜4周应监测血脂水平,并据此对剂量进行相应的调整。
⑥对纯合子高胆固醇血症患者,除非无法进行其他治疗,本品应作为其他降脂治疗措施(LDL血浆分离置换)基础上进行的治疗手段。
2.研究背景及作用机制
1研究背景
他汀类药物是20世纪80年代后期开发的羟甲戊二酰辅酶A(HMG-CoA)还原酶抑制剂类降血脂
药。
自从洛伐他汀问世后,先后又有辛伐他汀、普伐他汀、氟伐他汀、阿托伐他汀等用于临床,在降血脂药物中占据极为重要的地位[1]。
其中阿托伐他汀钙(立普妥)自2000年以来一直占据全
球畅销药第1位,受专利即将到期的影响,2009年全球市场销售额为123亿美元,比2008年的132.88亿美元有所下降。
尽管如此,作为唯一的1个年销售额连续多年超过百亿美元的药物,
随着专利的即将到期,其合成新工艺的优化再次引起科学
家们的关注。
氟伐他汀是第一个全合成的HMG-CoA还原酶抑制剂,而阿托伐他汀是第二个全
合成的他汀类药物,含有氟苯环和氮杂环,水溶性相对增大,脂溶性相对降低[2]。
2阿托伐他汀钙的作用机制
人体内大部分内源性胆固醇由肝脏合成,HMG-CoA还原酶是肝细胞合成胆固醇过程中的
限速酶,若该限速酶被抑制则能减少内源性胆固醇的合成[3]。
阿托伐他汀钙属他汀类药物,本
身及其代谢产物的化学结构与HMG-CoA还原酶相似,且与HMG-CoA还原酶的亲和力高,对该酶有竞争性抑制作用,因此能妨碍内源性胆固醇的合成⑷,从而有效治疗高血脂症。
3.合成路线
1.消旋体拆分法
在20世纪80年代Parke2Davis小组首次开发了1的化学小规模合成,先是合成外消旋体,然后再对非对映体进行拆分(Scheme1)。
利用3-氨基-1-丙醛缩乙二醇与a-溴代酯
(2)
烷烃化得到a-氨基酸酯(3)。
3与异丁酰氯反应,再水解得到a-酰胺酸(4)。
4在过量的3-苯基丙炔酰苯胺(5)存在下,在乙酐中加热得到[3+2]环加成产物6。
6通过一锅煮得到二乙基缩醛化合物,再酸性水解得到醛7。
在Weiler[4]条件下,7与乙酰乙酸乙酯的二价阴离子反应引入3-羟基戊内酯所需的碳和5-位羟基得8。
然后,8在低温(-78C)下用
n-Bu3B和NaBH4经Narasaka和Pai的非对映选择性还原法得到3,52-二醇(9,cis:
trans
=9:
1)o9经水解,然后在甲苯中回流内酯化得到1的内酯10(trans:
cis=9:
1)。
10经重结晶一次可使其trans:
cis>
97:
3。
10的拆分见Scheme2。
先将10制备成非对映的(R)-a-甲基苄酰胺(11),11经HPLC分离后水解,并重新内酯化得到94%光学纯的(+)-12。
这是一个分离一对类似的HMG抑制
剂的有效方法。
消旋体拆分法由于必须通过拆分才能得到最终产物,因此,产率不高。
从原子
经济性考虑是不可取的。
其意义在于建立了合成1对映体的方法,给后续对映选择性合成方法
的开辟指明了方向。
F
2.非对映选择性醇醛缩合法
Braun等利用7与(S)-(+)-2-乙酸基-1,1,2-三苯基乙醇进行非对映选择性
醇醛缩合得到手性化合物13,产率60%,e.e.值达到97%。
13再经过酯交换,选择性还原后关环得到含手性羟基的内酯(+)-12,e.e.值在99%以上。
(+)-12在碱性条件下水解开环后再成盐制得1(Scheme3)。
Scheme3为第一条手性合成1的路径,成功得到了克级的1。
虽然该路线存在明显的缺陷,如反应中涉及多个低温反应,最后的转化率低,难以实现工业化等,但该路线的建立却为后续合成方法的建立开辟了新的视野。
3.Paal-Knorr反应合成法
Parke-Davis小组利用异抗坏血酸(15)在碱性下开环、溴代酯化,再经催化氢化得到中间体(S)-4-溴-3-羟基丁酸甲酯(18)。
18的羟基经保护后,再经亲核取代得到腈19。
19用氢氧化钠水解,并通过羰基二咪唑的作用增长碳链,然后与丙二酸叔丁基单酯的镁盐反应并酸化,脱保护得到S-羟基-B-酮酸酯(21)o21经
NaBH4和Et2B0M选择性还原得到顺-1,3-二醇(22)。
22经叉丙酮保护后得到了晶
型良好的腈23,产率65%,非对映选择性为100:
1,经一次重结晶可以提高至350:
1o
23用含钼的Raney-Ni催化还原得到手性边链24,它的对映体过量百分率高达99.55%。
24与M4(Chart2)发生经典的Paal-Knorr反应得到了全取代的吡咯环25,
25用前述类似方法水解,脱保护,成盐制得1(Schemed),两步收率为45%主环与侧链缩合是经典的paal-knorr缩合,溶剂对此缩合收率有较大影响,国外对溶剂进行了选择,认为三氟甲苯最佳。
另可以先将24在甲苯:
庚烷:
四氢呋喃(1:
4:
1)中特戊酸成盐,成盐收率为87%再与M4(24与M4的摩尔比为1.3:
1)缩合得到25.,收率达70%25水解后成钙盐收率为81.3%,两步收率为57%
OH
HO
另外,Parke-Davis等还设计了另外两条合成手性边链的路线。
一是利用手性中间体18在羟基未经保护的情况下与乙酸叔丁酯在(i-Pr)2NLi的作用下直接得到了S-羟基-B-酮酸酯21(Scheme5),产率达到75%〜80%,进一步缩短了步骤,
提高了产率,已经达到公斤级的规模。
除了以抗坏血酸为原料合成18外,
Parke-Davis等还利用商业可得的手性环氧化合物经金属氰化物开环来合成
(Scheme6)。
二是利用商业可得的手性醇26(合成多个他汀类化合物的关键中间体)转化得到4-卤代或4-硝基苯磺酸酯(27),27再用氰化钠取代得到23,产率
高达80%(Scheme7)。
另外,Parke-Davis小组还利用手性中间体羟基腈酸酯(28)与N,N-二取代
的乙酰胺在LDA的作用下增链得到S-羟基-B-酮酰胺(29),29经还原,羟基保护,催化氢化得到手性酰胺边链32,32与M4发生Paal2Knorr反应得到吡咯环33,33经脱保护,水解成盐制得1(Scheme8)。
至于M4的合成,Parke-Davis小组利用商业可得的异丁酰乙酰苯胺(34)与
苯甲醛在B-氨基丙酸和乙酸存在下缩合得到a,B不饱和酮54,产率85%。
54和对氟苯甲醛在无水条件下以N-乙基噻唑为催化剂发生Stetter反应得到得M4,产率80%(Scheme13)。
由于该方法反应条件温和,原料简单,产率高,因此成为合成M4的经典方法。
II
4.环加成法
2004年辉端公司的Nelson等以7为原料,在手性Lewis酸的作用下发生不对
称羟醛缩合得手性醇35,产率达95%,e.e.值也达到94.4%。
35与丙烯酰氯酯化
得36,36在Grubb催化剂的作用下关环形成不饱和的内酯环37,37与苄醇发生
选择性的Michael加成反应,再水解就得到含有羟基的手性内酯38,e.e.值达
94.4%,产率为72%。
38再经过碱性水解,成盐得到1(Scheme10)
B
环加成法另辟蹊径得到了1,产率和对映选择性都得到了保证,虽然仍有一步
低温反应,但是大大缩短了反应步骤,特别是增链的过程完全摆脱了前面提及的几
种方法,开辟了新的途径。
但是该方法也有明显的不足,其操作复杂,工业化实施
困难
5双羰基不对称还原法
2005年,Bulter等又提出了一系列合成1的路线,他们以a-氰基乙酸甲酯
(39)为原料,与吗啉形成酰胺40,40经催化氢化得到伯胺41,41与M4发生
Paal2Knorr反应形成吡咯环42,42与乙酰乙酸叔丁酯缩合增链形成一个多羰基化合物43,43在钉的手性催化剂的催化下发生不对称氢化得到手性羟基化合物44,
44内酯化后再与苄醇发生选择性Michael加成、水解得到含手性羟基的内酯38,
38经水解、成盐得到1(Scheme11)。
双羰基不对称还原法与环加成法同样缩短了反应步骤提高了产率,但是我们也
看到其中43的不对称催化氢化是反应的关键,昂贵的手性催化剂给工业化生产带来了困难
3.专利情况
作为全球第一的药物,其合成专利极为丰富,而paal-knorr最为主要合成方法,其关键中间体ATS-9与M4的合成方法便成为大家争相研究的的对象,现选取几例作简要介绍。
3.1边链中间体ATS-9的其他合成法
由于手性边链包含了1的所有手性元素,成为合成1的关键。
2001年,姜标小组以B-邻苯二甲酰亚胺基丙醛(45)为原料在盐酸羟胺作用下发生羟胺化反应得到3-N-邻苯二甲酰亚胺基-丙肟(46)。
46经N-氯代邻苯二甲酰亚胺或NaOCI氯化得47,47再与3-丁酰酸酯发生1,3-偶极加成成功地增长碳链得到48,48经水解后,在马钱子碱或奎宁等手性碱的作用下拆分得到手性中间体49,e.e.值在95%以上。
50经酯化,催化氢化开环,选择性还原后得到1,3-顺式二醇51。
51保护后经酸解或肼解后成功得到1的手性边链24(Scheme12)。
o
45
2004年,Mark小组利用环氧氯丙烷(52)开环后的产物53,在腈水解酶的作
用下成功地得到了e.e.值高达97%的手性中间体羟基腈酸(54)。
54重结晶一次后e.e.值可达99.6%,然后酯化得到18(Scheme13),进一步拓展了醇醛缩合法
2002年,韩国三星精细化学公司又设计了合成手性边链中间体的路线。
他们以商业可得的(S)-4-溴代或氯代-3-羟基丁腈(55a或55b)为原料,将羟基保护后在羰基二咪唑和Meldrum酸的作用下增链,再经取代后得了含有一个手性碳的边链59。
再经过选择性还原,脱保护后就得到了含有两个手性碳的边链腈22(Scheme14)。
Br
2004年,韩国LG化学公司同样以55a或55b为原料与a-溴代乙酸叔丁酯发生
Blaise反应,然后在酸性条件下水解得到另一个合成边链的中间体60,60再经选
择性还原得到顺式二醇61,61的双羟基经保护后,再发生取代反应得到23(Scheme
15)。
3.2母核M4的其他合成方法
2003年,Schoning等以苯乙酰氯和氟苯为原料,在AICI3的催化下发生傅-
克反应得到化合物63,产率高达90%。
63再经过溴代得到几乎等当量的a-溴代酮
64。
64与34在碱性条件下缩合以80%的产率得到M4(Scheme16)。
2009年,沈阳医科大学的李慧敏等以价廉易得的丙二酸为原料,在浓硫酸的催化下醋酸酐做脱水剂和丙酮缩合制得麦尔德姆酸(65),收率53.3%,,65与异丁酰氯(66)以吡啶为脱羧剂反应生成酰基化的麦尔德姆酸(66),66再与苯胺反应制得异丁酰乙酰苯胺(68)两步收率可达88.8%。
(scheme17)
将苯乙酸与二氯亚砜反应制取苯乙酰氯(69),收率97.1%,69与氟苯以氯化铝为催化剂缩合得4-氟苯基苯乙酮(70),此部产率为74.5%,将70溶于冰醋酸,逐滴加溴水使其溴化,得2-溴-1-4-氟苯基苯乙酮(71),71与68在碳酸钾与丙酮存在条件下反应即可制得M4,产率达97.1%
各种边链ATS-9和M4的合成方法的建立大大丰富了阿托伐他汀钙的合成路
线,为寻找一条简单、经济、环境友好和易于工业化的合成路线创造了条件。
并
且,许多新的合成方法的应用也为其他类似化合物的合成开辟了新的途径,大大缩短了反应步骤。
4.结论
尽管报道了多种合成阿托伐他汀钙的方法,但经典的Paal-Knorr法是目前的主流合成方
法。
该方法的关键在于手性侧链ATS和中间体M-4的合成,因此,发展高效的构建M-4的
合成方法以及手性侧链ATS的不对称合成仍将是未来研究的热点。
另外,其他的合成方法也为研究者合成阿托伐他汀钙提供了不同选择。
 升级会员
升级会员 