第一类医疗器械备案申请材料.docx
《第一类医疗器械备案申请材料.docx》由会员分享,可在线阅读,更多相关《第一类医疗器械备案申请材料.docx(8页珍藏版)》请在冰豆网上搜索。
第一类医疗器械备案申请材料
第一类医疗器械备案申请材料
********器械生产企业
****年**月**日
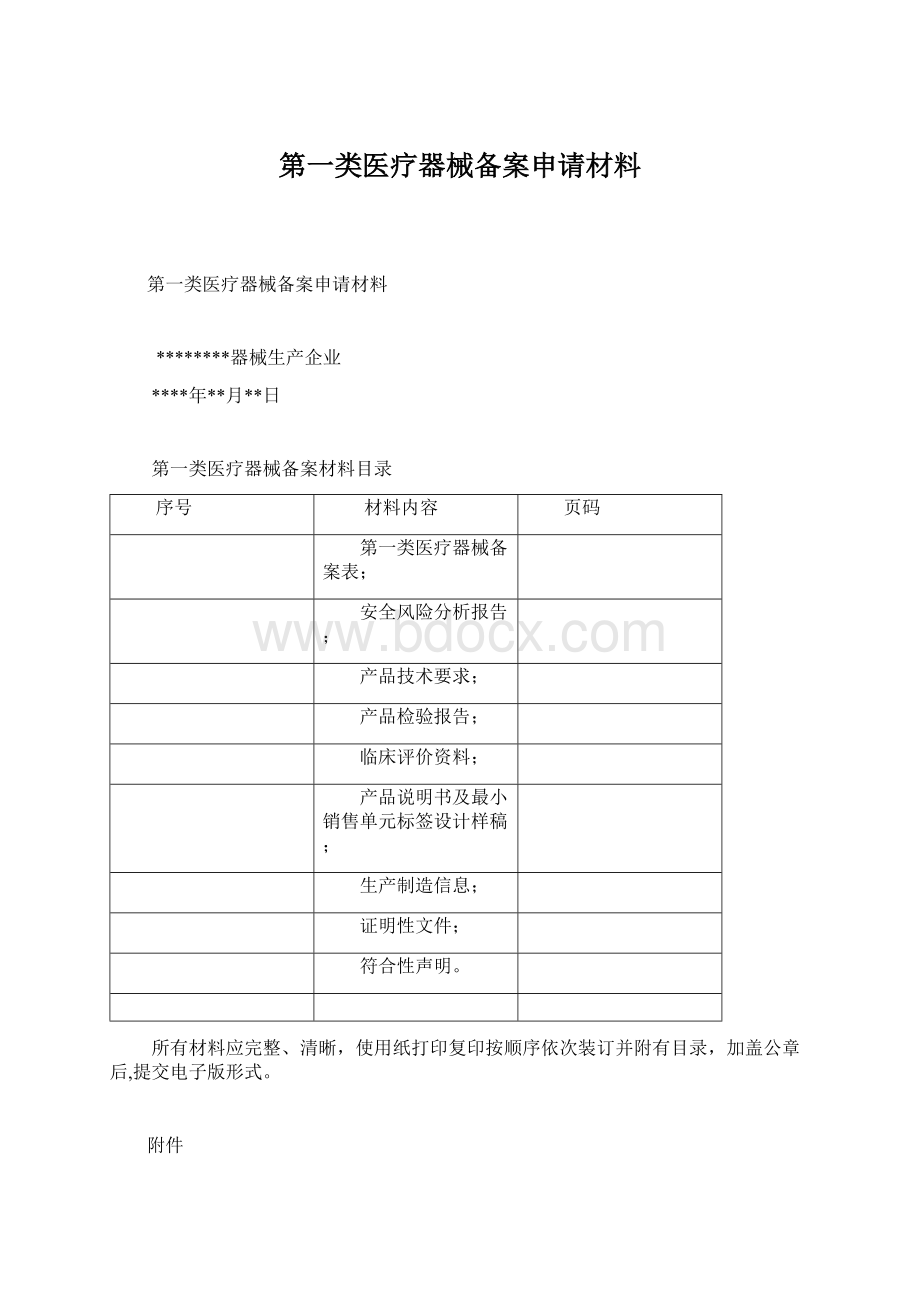
第一类医疗器械备案材料目录
序号
材料内容
页码
第一类医疗器械备案表;
安全风险分析报告;
产品技术要求;
产品检验报告;
临床评价资料;
产品说明书及最小销售单元标签设计样稿;
生产制造信息;
证明性文件;
符合性声明。
所有材料应完整、清晰,使用纸打印复印按顺序依次装订并附有目录,加盖公章后,提交电子版形式。
附件
备案资料形式要求
一、备案资料完整齐备。
备案表填写完整。
二、各项文件除证明性文件外均应以中文形式提供。
如证明性文件为外文形式还应提供中文译本。
根据外文资料翻译的申报资料,应同时提供原文。
三、境内产品备案资料如无特殊说明的,应由备案人签章。
“签章”是指:
备案人盖章,或者其法定代表人、负责人签名加企业盖章。
所盖章必须是备案人公章,不得使用注册专用章。
四、进口产品备案资料如无特别说明,原文资料均应为原件,并由备案人签章,中文文本由代理人签章。
原文资料“签章”是指:
备案人的法定代表人、负责人签名,或者签名加组织机构盖章,并且应当提交由备案人所在地公证机构出具的公证件;中文资料“签章”是指:
代理人的组织机构盖章,或者其法定代表人、负责人签名加组织机构盖章。
五、备案资料应有所提交资料目录,包括整个申报资料的级和级标题,并以表格形式说明每项的卷和页码。
附件
第一类医疗器械备案资料要求及说明
(一)第一类医疗器械备案表
(二)安全风险分析报告
医疗器械应按照《医疗器械风险管理对医疗器械的应用》的有关要求编制,主要包括医疗器械预期用途和与安全性有关特征的判定、危害的判定、估计每个危害处境的风险;对每个已判定的危害处境,评价和决定是否需要降低风险;风险控制措施的实施和验证结果,必要时应引用检测和评价性报告;任何一个或多个剩余风险的可接受性评定等,形成风险管理报告。
体外诊断试剂应对产品寿命周期的各个环节,从预期用途、可能的使用错误、与安全性有关的特征、已知和可预见的危害等方面的判定及对患者风险的估计进行风险分析、风险评价及相应的风险控制的基础上,形成风险管理报告。
(三)产品技术要求
产品技术要求应按照《医疗器械产品技术要求编写指导原则》编制。
(四)产品检验报告
产品检验报告应为产品全性能自检报告或委托检验报告,检验的产品应当具有典型性。
(五)临床评价资料
.详述产品预期用途,包括产品所提供的功能,并可描述其适用的医疗阶段(如治疗后的监测、康复等),目标用户及其操作该产品应具备的技能知识培训;预期与其组合使用的器械。
.详述产品预期使用环境,包括该产品预期使用的地点如医院、医疗临床实验室、救护车、家庭等,以及可能会影响其安全性和有效性的环境条件(如温度、湿度、功率、压力、移动等)。
.详述产品适用人群,包括目标患者人群的信息(如成人、儿童或新生儿),患者选择标准的信息,以及使用过程中需要监测的参数、考虑的因素。
.详述产品禁忌症,如适用,应明确说明该器械禁止使用的疾病或情况。
.已上市同类产品临床使用情况的比对说明。
.同类产品不良事件情况说明。
(六)产品说明书及最小销售单元标签设计样稿
医疗器械应符合相应法规规定。
进口医疗器械产品应提交境外政府主管部门批准或者认可的说明书原文及其中文译本。
体外诊断试剂产品应按照《体外诊断试剂说明书编写指导原则》的有关要求,并参考有关技术指导原则编写产品说明书。
进口体外诊断试剂产品应提交境外政府主管部门批准或者认可的说明书原文及其中文译本。
(七)生产制造信息
对生产过程相关情况的概述。
无源医疗器械应明确产品生产加工工艺,注明关键工艺和特殊工艺。
有源医疗器械应提供产品生产工艺过程的描述性资料,可采用流程图的形式,是生产过程的概述。
体外诊断试剂应概述主要生产工艺,包括:
固相载体、显色系统等的描述及确定依据,反应体系包括样本采集及处理、样本要求、样本用量、试剂用量、反应条件、校准方法(如果需要)、质控方法等。
应概述研制、生产场地的实际情况。
(八)证明性文件
.境内备案人提供:
企业营业执照复印件、组织机构代码证复印件。
.境外备案人提供:
()境外备案人企业资格证明文件。
()境外备案人注册地或生产地址所在国家(地区)医疗器械主管部门出具的允许产品上市销售的证明文件。
备案人注册地或生产地址所在国家(地区)不把该产品作为医疗器械管理的,备案人需提供相关证明文件,包括备案人注册地或生产地址所在国家(地区)准许该产品合法上市销售的证明文件。
如该证明文件为复印件,应经当地公证机关公证。
()境外备案人在中国境内指定代理人的委托书、代理人承诺书及营业执照副本复印件或者机构登记证明复印件。
(九)符合性声明
.声明符合医疗器械备案相关要求;
.声明本产品符合第一类医疗器械产品目录或相应体外诊断试剂分类子目录的有关内容;
.声明本产品符合现行国家标准、行业标准并提供符合标准的清单;
.声明所提交备案资料的真实性。
材料
备案号:
第一类医疗器械备案表
(参考格式)
产品名称(产品分类名称):
备案人:
食品药品监督管理局制
(国家食品药品监督管理总局制)
填表说明
1.本表用于进口和境内第一类医疗器械、体外诊断试剂备案。
2.要求填写的栏目内容应使用中文、打印完整、清楚、不得空白,无相关内容处应填写“∕”。
因备案表格式所限而无法填写完整时,请另附附件。
3.备案时应一并提交含有备案表内容(含附件)的电子文档(形式)。
4.境内医疗器械、体外诊断试剂只填写备案人名称、注册地址和生产地址中文栏。
进口医疗器械、体外诊断试剂备案人名称、注册地址和生产地址中文栏自行选择填写。
进口医疗器械产品名称(体外诊断试剂为产品分类名称,以下同)中文栏必填。
5.如系统支持,则进口医疗器械产品名称、备案人名称、注册地址和生产地址原文栏必填,原文填写内容应与备案人注册地址或生产地址所在国家(地区)医疗器械主管部门出具的允许产品上市销售的证明文件中载明内容和文种一致。
6.境内医疗器械备案人应填写组织机构代码。
7.进口医疗器械产品名称、备案人名称、注册地址和生产地址英文栏必填。
如原文非英文,英文内容必须与原文一致。
8.所填写各项内容应与所提交备案材料内容相对应。
9.产品类别及分类编码应根据医疗器械分类规则和医疗器械分类目录、第一类医疗器械产品目录、第一类体外诊断试剂分类子目录等相关文件填写。
10.备案人、代理人注册地址栏填写备案人和代理人企业营业执照等相关证明性文件上载明的注册地址。
11.备案人、代理人所在地系指备案人和代理人注册地址所在国家(地区)或省(区、市)。
12.如有其他需要特别加以说明的问题,请在本表“其他需要说明的问题”栏中说明。
注:
填表前,请详细阅读填表说明
产品名称
(产品分类名称)
中
文
原
文
英
文
分类编码
结构特征
有源□无源□体外诊断试剂□
型号规格
(包装规格)
产品描述
(主要组成成分)
预期用途
产品有效期(体外
诊断试剂适用)
备案人
名称
中文
原文
英文
注册地址
中文
原文
英文
联系人
电话
传真
电子邮箱
邮编
备案人
所在地
组织机构代码
生产地址
中文
原文
英文
代理人
名称
注册地址
邮编
联系人
电话
传真
电子信箱
代理人
所在地
应附资料
1.产品风险分析资料□
2.产品技术要求□
3.产品检验报告□
4.临床评价资料□
5.生产制造信息□
6.产品说明书及最小销售单元标签设计样稿□
7.证明性文件□
8.符合性声明□
其他需要说明的问题
备案人代理人(签章)
日期:
年月日
材料
安全风险分析报告。
材料
产品技术要求。
材料
产品检验报告。
材料
临床评价资料。
材料
产品说明书及最小销售单元标签设计样稿。
材料
生产制造信息。
材料
证明性文件。
材料
符合性声明。
 升级会员
升级会员 