药理学期末总复习汇总.docx
《药理学期末总复习汇总.docx》由会员分享,可在线阅读,更多相关《药理学期末总复习汇总.docx(83页珍藏版)》请在冰豆网上搜索。
药理学期末总复习汇总
药理学
期末总复习
总论、药物代谢动力学、药物效应动力学
药物:
是指可以改变或查明机体的生理功能和病理状态,用以预防、诊断和治疗疾病的物质。
药理学是研究药物与机体(含病原体)相互作用和作用规律的学科。
离子障:
分子型药物疏水而亲脂,易通过细胞膜;离子型药物极性高,不易通过细胞膜脂质层,这种现象称为离子障。
药物过细胞膜的方式包括滤过、简单扩散、载体转运、膜动转运。
绝大多数是通过简单扩散。
药物的体内过程要点:
给药途径有口服、吸入、局部用药、舌下给药、注射给药
口服是最常用的给药途径,给药方便。
大多数药物能充分吸收。
大多数药物在胃肠道内以简单扩散方式被吸收。
首关消除:
从胃肠道吸收如门静脉系统的药物在到达全身血液循环之前必先通过肝脏,如果肝脏对其代谢能力很强,或由胆汁排泄的量很大,则进入全身血液循环内的有效药物量明显减少,这种作用称为首关消除。
直肠给药可在一定程度上避免首关消除。
舌下给药可在很大程度上避免首关消除。
血管注射避开了吸收屏障而直接入血,不存在吸收过程。
药物肌内注射一般比皮下注射吸收快。
分布:
药物吸收后从血液循环到达机体各个器官和组织的过程称为分布。
分布的影响因素:
脂溶度、毛细血管通透性、器官和组织的血流量、与血浆蛋白和组织蛋白的结合能力,药物的pKa和局部的pH、药物载体转运蛋白的数量和功能状态、特殊组织膜的屏障作用等。
结合型药物不能跨膜转运,是药物在血液中的一种暂时贮存形式。
药物与血浆蛋白结合的特异性低,与相同血浆蛋白结合的药物之间可发生竞争性置换的相互作用。
屏障:
血脑屏障;只有脂溶性高的药物才能以简单扩散的方式通过血脑屏障。
胎盘屏障;胎盘对药物的转运并无屏障作用,几乎作用的药物都能穿透胎盘进入胎儿的体内。
血眼屏障;脂溶性或小分子药物容易透过血眼屏障。
代谢:
药物在体内经酶或其他作用使药物的化学结构发生改变,这一过程称为代谢或生物转化。
肝脏是最主要的代谢器官。
药物的生物转化和排泄速度决定其作用持续时间的长短。
药物代谢使得药物的极性变大,易于代谢出体外。
自身诱导:
有些药物本身就是其所诱导的药物代谢酶的底物,因此在反复应用后,药物代谢酶的活性增高,药物自身代谢也加快,这一作用称自身诱导。
肾脏排泄分为肾小球滤过、肾小管分泌和肾小管重吸收几个部分。
试述体液pH值改变如何影响弱酸性和弱碱性药物在体内的吸收、分布和排泄。
1)答:
体液pH值的改变主要通过改变药物的解离、非离子分子比值而影响弱酸性和弱碱性药物的吸收。
弱碱性药物在碱性环境的小肠内解离度小,主要以非解离型存在,故在小肠吸收较多。
同理,弱酸性药物在酸性环境的胃内较易吸收。
2)体液pH值改变可影响药物在体内的分布。
若提高血液的pH值,可使弱酸性药物在细胞外液增多,使弱碱性药物向细胞内转移增多。
但巴比妥类药物中毒时,静滴碳酸氢盐使血液和尿液碱化,毒物不易进入细胞内,尤其不易进入脑细胞,同时又促使毒物自尿排泄,起到解毒目的。
3)尿液pH值改变决定药物排泄。
弱酸性药物在碱性尿中解离多,重吸收少,排泄多。
反之,在酸性尿中解离少,重吸收多,排泄少。
弱碱性药物在酸性尿中重吸收少,排泄多,在碱性尿中,重吸收多,排泄少。
简述一级动力学消除的特点。
答:
1)一级消除动力学是体内药物按恒定比例消除,在单位时间内的消除量与血浆药物浓度成正比。
2)半衰期恒定,不因血药浓度的高低而改变。
3)单次给药后在体内的消除时间约为5个半衰期。
4)按固定剂量、固定间隔时间给药、或恒速静脉滴注,经4-5个半衰期基本能达到稳态血药浓度。
5)大多数药物在体内按照一级动力学消除。
6)药-时曲线在常规坐标图上作图呈曲线,在半对数坐标图上呈直线。
简述零级消除动力学的特点。
1)答:
零级消除动力学是药物在体内以恒定的速率消除,即不论血浆药物浓度高低,单位时间内消除的药物量不变。
2)药物消除速度与初始浓度无关。
3)半衰期不是恒定值,和血浆药物的初始浓度成正比,给药剂量越大,半衰期越长。
4)只有少数药物的消除属于零级消除动力学,药物容易出现蓄积中毒。
5)药-时曲线在常规坐标图上呈曲线,在半对数坐标图上呈直线。
混合消除动力学:
一些药物在体内可表现为混合消除动力学,即在低浓度或低剂量时,按一级动力学消除,达到一定高浓度或高剂量时,因消除能力饱和,单位时间内消除的药物量不再改变,按零级动力学消除,如苯妥英钠,水杨酸等。
药物消除半衰期是血浆药物浓度下降一半所需要的时间,长短可反映体内药物消除速度。
多次给药后药物达到稳态浓度的时间仅取决于药物的消除半衰期。
一般来说,药物在剂量和给药间隔时间不变时,经4-5个半衰期可分别达到稳态浓度的94%和97%。
表观分布容积:
当血浆和组织内药物分布达到平衡时,体内药物按血浆药物浓度在体内分布所需体液容积。
表观分布容积小,与血浆蛋白结合一般较多,较集中于血浆。
表观分布容积大,与血浆蛋白结合一般较少,主要分布于血浆外的组织。
生物利用度是什么?
试比较相对利用度跟绝对利用度。
答:
生物利用度是指药物经血管外途径给药后吸收进入全身血液循环的相对量。
药物在体内的量可用AUC来表示,因为静脉注射时的生物利用度应为100%,因此如以血管外给药(如口服)的AUC和以静脉注射的AUC进行比较,可得该药的绝对生物利用度。
对同一血管外给药途径的某一种药物制剂(如不同剂型、不同药厂生产的相同剂型、同一药厂生产的同一品种的不同批号等)的AUC与相同的标准制剂进行比较,可得相对利用度。
药物作用是指药物对机体的初始作用,是动因。
药物效应是药物作用的结果,是机体反应的表现。
药理效应是机体器官原有功能水平的改变,功能提高称为兴奋,功能降低称为抑制。
药物的作用有选择性,有些药物可影响机体的多种功能,有些药物只影响机体的一种功能,前者选择性低,后者选择性高。
不良反应:
凡与用药目的无关,并为患者带来不适或痛苦的反应统称为药物不良反应,一般情况下是可以预知的,但不一定是能够避免的。
分为:
1.副反应:
由于选择性低,药理效应涉及多个器官,当某一效应用作治疗目的时,其他效应就成为副反应。
副反应是在治疗剂量下发生的,是药物本身固有的作用,多数较轻并可以预料。
2.毒性反应:
毒性反应是指在剂量过大或药物在体内蓄积过多时发生的危害性反应,一般比较严重。
毒性反应一般是可以预知的,应该避免发生。
急性毒性多损害循环,呼吸和神经系统功能。
慢性毒性多损害肝、肾、骨髓、内分泌等功能。
致癌和致畸变和致突变反应也属于慢性毒性范畴。
3.后遗效应:
是指停药后血药浓度已降至阈浓度以下时残存的药理效应。
4.停药反应:
是指突然停药后原有疾病加剧,又称回跃反应。
5.变态反应:
是一类免疫反应。
常见于过敏体质患者,反应性质与药物原有效应无关,用药理性拮抗药解救无效,反应的严重程度差异很大,与剂量无关。
6.特异质反应:
少数特异体质患者对某些药物反应特别敏感,反应性质也可能与常人不同,但与药物固有的药理作用基本一致,反应程度与剂量成比例,药理性拮抗药救治可能有效。
药理效应按性质可以分为量反应和质反应。
效应的强弱呈连续增减的变化,可用具体数量或最大反应的百分率表示者称为量反应。
以药物的剂量或浓度为横坐标,以效应浓度为纵坐标作图,可获得直方双曲线,如将药物浓度改用对数值作图则呈典型的对称S形曲线,这就是通常所称量反应的量-效曲线。
最小有效量或最低有效浓度:
刚能引起效应的最小药量或最小药物浓度,亦称阈剂量或阈浓度。
最大效应:
随着剂量或浓度的增加,效应也增加,当效应增加到一定程度后,若继续增加药物浓度或剂量而其效应不再继续增强,这一药理效应的极限称为最大效应,也称效能。
半最大效应浓度(EC50)是指能引起50%最大效应的浓度。
效价强度:
是指能引起等效反应(一般采用50%效应量)的相对浓度或剂量,其值越小则强度越大。
如果药理效应不是随着药物剂量或浓度的增减呈持续性量的变化,而表现为反应性质的变化,则称为质反应。
如果按照药物浓度或剂量的区段出现阳性反应的频率作图得到呈常态分布曲线。
如果按照剂量增加的累计阳性反应百分率作图,则可得到典型的S形量效曲线。
半数有效量(ED50):
能引起50%的实验动物出现阳性反应时的药物剂量,如效应为死亡,则称为半数致死量(LD50)。
通常将药物的LD50/ED50的比值称为治疗指数,用以表示药物的安全性。
治疗指数大的药物相对较治疗指数小的药物安全。
还可用1%致死量(LD1)和99%有效量(ED99)的比值或5%致死量(LD5)与95%有效量(ED95)之间的距离来衡量药物的安全性。
受体:
受体是一类介导细胞信号转导的功能蛋白质,能识别周围环境中某种微量化学物质,首先与之结合,并通过中介的信息放大系统,触发后续的生理反应或药理反应。
体内能与受体特异性结合的物质称为配体,也称第一信使。
受体的特性:
灵敏性、特异性、饱和性、可逆性、多样性
药物和受体结合,不仅需要亲和力,而且还要有内在活性。
所谓的内在活性是指药物与受体结合后产生效应的能力。
受体激动药:
为既有亲和力又有内在活性的药物,它们能与受体结合后并激动受体而产生效应。
依其内在活性大小又可分为完全激动药和部分激动药。
前者具有较强亲和力和较强的内在活性,后者有较强亲和力,但内在活性不强。
受体拮抗药:
能与受体结合,具有较强亲和力而无内在活性的药物。
他们本身不产生作用,但因占据受体而拮抗激动药的效应。
少数拮抗药以拮抗作用为主,同时尚有较弱的内在活性,故有较弱的激动受体作用。
pD2:
即亲和力指数,受体激动药和受体的亲和力大小可用pD2来表示,是受体激动药半最大效应浓度的负对数值,pD2大者亲和力大。
pA2:
即拮抗参数,即当激动药与拮抗药合用时,若2倍浓度的激动药所产生的效应恰好等于未加入拮抗药时激动药所产生的效应,则所加入的拮抗药的摩尔浓度的负对数值即称之为pA2,pA2值越大,拮抗作用越强。
受体的调节:
受体脱敏:
是指在长期使用一种激动药后,组织或细胞对激动药的敏感性和反应性下降。
如仅对一种类型的受体激动药的反应性下降,而对其他类型受体激动药的反应性不变,则称为激动药特异性脱敏,若组织或细胞对一种类型激动药脱敏,对其他类型受体激动药也不敏感,则称为激动药非特异性脱敏。
受体增敏:
是与受体脱敏相反的一种现象,可因受体激动药水平降低或长期应用拮抗药而造成。
若受体脱敏和增敏只涉及受体密度的变化,则分别称之为下调或上调。
耐受性:
耐受性为机体在连续多次用药后对药物的反应性降低。
增加剂量可恢复反应,停药后耐受性可消失。
交叉耐受性:
是对一种药物产生耐受性后,在应用同一类药物(即使是第一次使用)时也会产生耐受性。
耐药性:
是指病原体或肿瘤细胞对反复应用的化学治疗药物的敏感性降低,也称抗药性。
依赖性:
长期应用某种药物后,机体对这种药物产生生理性或精神性的依赖和需求。
长期用药的患者停药时必须逐渐减量至停药,可避免停药综合征的发生。
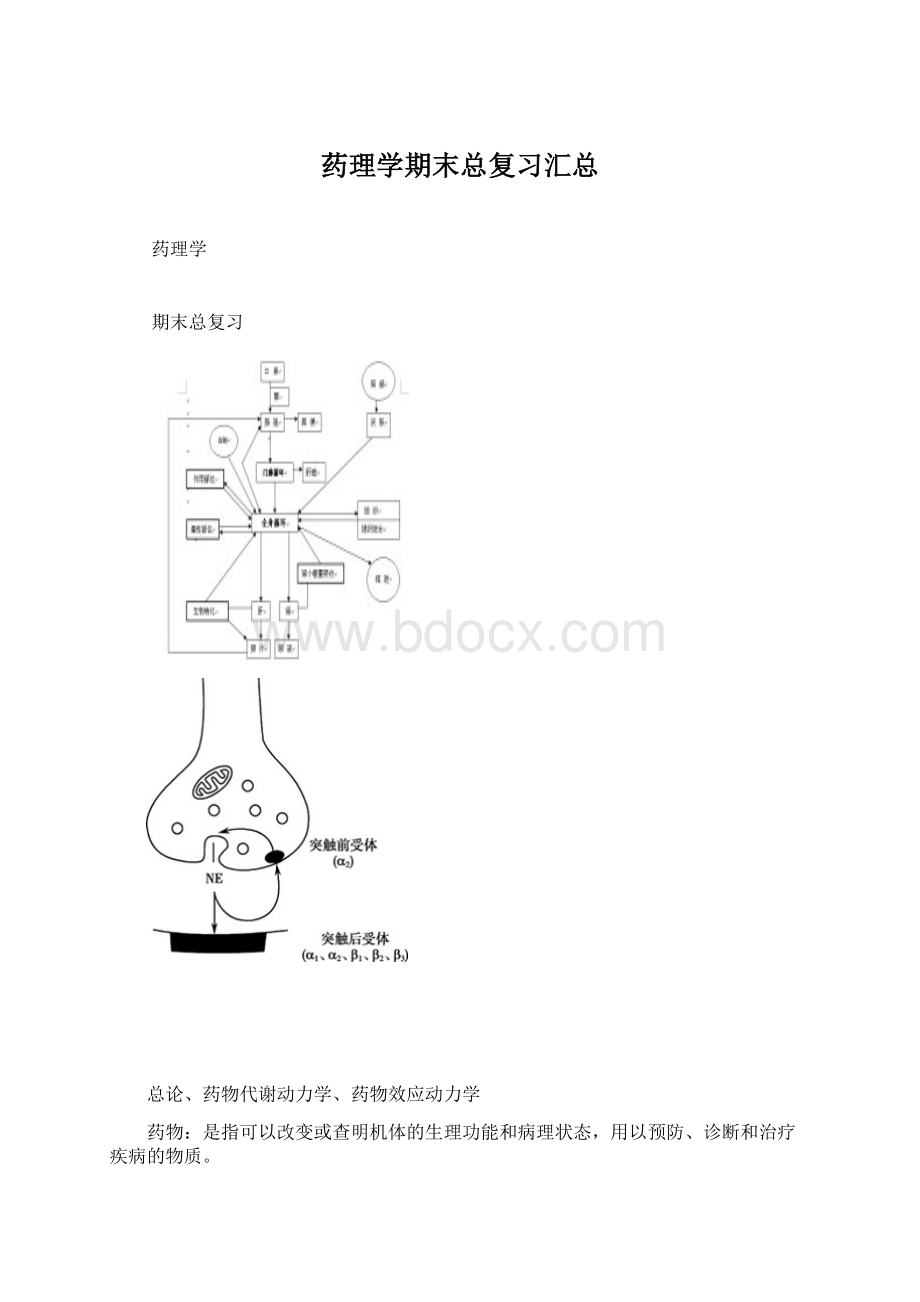
传出神经系统的药理
传出神经根据其末梢释放的递质不同,可分为以乙酰胆碱为递质的胆碱能神经和主要以去甲肾上腺素为递质的去甲肾上腺素能神经。
胆碱能神经主要包括全部交感神经和副交感神经的节前纤维、运动神经、全部副交感神经的节后纤维和极少数交感神经的节后纤维(支配汗腺分泌和骨骼肌血管舒张神经)。
去甲肾上腺素能神经则包括几乎全部交感神经节后纤维。
能与ACh特异结合的受体称为胆碱能受体。
根据药理学特性,胆碱能受体可以分为两类,一类能与天然植物中的毒蕈碱结合,称为毒蕈碱受体,简称M受体;另一类能与天然植物中的烟碱结合,称为烟碱受体,简称N受体。
两类受体与ACh结合后产生不同的生物学效应。
M受体有五种亚型,他们均为G蛋白偶联受体。
N受体有两种亚型,都是离子通道受体。
M受体激活后可产生一系列自主神经效应,包括心脏活动抑制,支气管和胃肠平滑肌、膀胱逼尿肌、虹膜环形肌收缩,消化腺、汗腺分泌增加和骨骼肌血管舒张等。
这些作用统称为毒蕈碱样作用,简称M样作用。
小剂量ACh作用于N受体后,能兴奋自主神经节节后神经元,也能收缩骨骼肌;而大剂量ACh作用于N受体后,则可阻断自主神经节的突触传递,这些作用统称为烟碱样作用,简称N样作用。
M胆碱受体激动药
乙酰胆碱(性质不稳定,极易被AChE水解,且作用广泛,选择性差)
1.心血管系统:
1)舒张血管(内皮依赖性舒张因子,并抑制NA释放)
2)减弱心肌收缩力3)减慢心率4)减慢房室结和普肯耶纤维传导5)缩短心房不应期
2.胃肠道:
兴奋胃肠道平滑肌,使其收缩幅度、张力和蠕动增加,能促进胃、肠分泌。
3.泌尿道:
使泌尿道平滑肌蠕动增加,膀胱逼尿肌收缩,膀胱三角区和外括约肌舒张,促进排空。
4.腺体:
使泪腺、气管和支气管腺体、唾液腺、消化道腺体和汗腺分泌增加。
5.眼:
瞳孔缩小,降低眼内压,调节痉挛
6.神经节和骨骼肌:
可引起交感、副交感神经节兴奋和骨骼肌收缩。
N1胆碱受体激动能引起肾上腺素释放。
7.支气管:
ACh可使支气管收缩。
醋甲胆碱(水解速度较ACh慢,作用时间长)
对M受体有相对选择性,尤其对心血管系统作用明显。
临床上主要用于口腔黏膜干燥症。
禁忌症为支气管哮喘、冠状动脉缺血和溃疡病患者。
卡巴胆碱(性质稳定,不易被水解,作用时间长)
对膀胱和肠道作用明显,可用于术后腹气胀和尿潴留。
副作用多,主要用于局部滴眼治青光眼。
(仅用于皮下注射,禁用静脉注射给药)
贝胆碱(性质稳定,不易被水解)
对心血管作用弱,可兴奋胃肠道和泌尿道平滑肌。
临床可用于术后腹气胀、胃张力缺乏症和胃滞留等的治疗。
效果较卡巴胆碱好。
口服注射均有效。
禁忌症:
支气管哮喘,冠状动脉缺血和溃疡病。
生物碱类
毛果芸香碱(匹鲁卡品)
对眼和腺体作用较明显。
1.眼:
滴眼后可引起缩瞳、降低眼内压和调节痉挛等作用。
1)缩瞳:
本品激动瞳孔括约肌的M胆碱受体。
2)降低眼内压:
本品通过缩瞳作用使虹膜向中心拉动,虹膜根部变薄,使虹膜周围的前房角间隙扩大,房水易于进入巩膜静脉窦,使眼内压下降。
3)调节痉挛:
(睫状肌收缩,悬韧带松弛。
睫状肌舒张,睫状肌收缩)本品使环状肌收缩,睫状肌收缩,悬韧带放松,晶状体变凸,只能看近物。
2.腺体:
较大剂量的毛果芸香碱的皮下注射能明显增加汗腺和唾液腺的分泌,也可使泪腺、胃腺、胰腺、小肠腺体和呼吸道黏膜分泌增加。
临床应用:
1)治疗青光眼:
用药后使患者瞳孔缩小,前方角间隙扩大,房水回流通畅,眼内压下降。
(滴眼时应压迫内眦,避免药液流入鼻腔增加吸收而产生不良反应。
)
2)虹膜睫状体炎
3)治疗口干舌燥。
4)阿托品中毒的解救
抗胆碱酯酶药:
易逆性抗胆碱酯酶药:
新斯的明:
药理作用:
季铵类化合物,较少进入中枢。
可抑制AChE活性而发挥完全拟胆碱作用,即通过ACh兴奋M、N胆碱受体,尚能直接激动N2受体,对骨骼肌的兴奋作用较强,兴奋胃肠平滑肌的作用其次,对腺体、眼、心血管和支气管平滑肌作用弱。
1.眼:
缩瞳、降低眼内压、调节痉挛
2.胃肠道:
促进胃平滑肌收缩和增加胃酸分泌,对食管下段具有兴奋作用,能促进食管的蠕动,并增加其张力。
可促进小肠、大肠的活动、促进肠内容物排出。
3.骨骼肌神经肌肉接头:
不仅通过抑制神经肌肉接头的AChE,也有一定的直接兴奋作用。
治疗剂量,使得骨骼肌收缩力增强,大剂量时,发生肌纤维震颤,继而整个运动单位的肌束震颤。
4.低剂量增敏神经冲动所致的腺体分泌作用,较高剂量增加基础分泌率。
引起细支气管和输尿管平滑肌收缩,使后者的蠕动增加。
临床主要应用:
1.重症肌无力。
治疗时需反复给药。
2.腹气胀和尿潴留。
3.解毒:
用于竞争性神经肌肉阻滞药过量的解毒,也用于M胆碱受体阻断剂药物中毒的解救。
4.阵发性室上性心动过速。
禁忌症:
机械性肠或泌尿道梗阻患者。
其他常用易逆性抗AChE药
吡斯的明:
起效较缓慢,作用时间较长。
口服吸收较差,剂量较大。
主要用于重症肌无力,也可用于麻痹性肠梗阻和术后尿潴留。
依酚氯胺:
常用于诊断重症肌无力
安贝氯胺:
较持久,主要用于治疗重症肌无力
毒扁豆碱:
为叔胺类化合物,可进入中枢。
主要用途为局部用于治疗青光眼。
(滴眼时,应压迫内眦,以免药液流入鼻腔后吸收中毒。
)(1/10的毒扁豆碱为加兰他敏)
地美溴铵:
主要用于治疗青光眼。
(重症肌无力主要用新斯的明,吡斯的明和安贝氯胺。
竞争性神经肌肉阻滞药过量时的解毒,主要用新斯的明、依酚氯胺和加兰他敏,青光眼主要用毒扁豆碱和地美溴铵)
难逆性抗AChE药-有机磷酯类
中毒要点:
从呼吸道吸入,全身中毒症状可在数分钟内出现。
如经胃肠道或皮肤吸收,则中毒症状可出现不同程度的延缓。
瞳孔明显变小、视力模糊、泪腺、唾液腺、支气管和胃肠道腺体分泌增加,呼吸困难。
毒物从胃肠道摄入,表现为厌食、恶心、呕吐、腹痛、腹泻等。
毒物经过皮肤吸收中毒时,则首先出现于吸收部位最邻近区域出汗和肌束颤动。
有机磷酸酯类农药急性中毒,如何用药物解救?
简述其机理和注意点。
答:
首先应迅速清除毒物,以免继续吸收。
并及时使用解毒药物。
再进行对症治疗。
1)要联合用药:
阿托品属于M受体阻断剂,能迅速解除有机磷酸酯类中毒时的M样症状。
AChE复活药如氯解磷定不仅可以恢复AChE的活性还可以直接与体内游离的有机磷酸酯类结合并排出,阻止游离的毒物继续抑制AChE的活性。
氯解磷定可明显减轻N样症状,对骨骼肌痉挛的抑制作用最为明显,能迅速抑制肌束颤动,对中枢神经系统的中毒症状也有一定改善作用。
2)要注意尽早用药:
阿托品应尽量早期使用。
磷酰化胆碱酯酶易老化,故AChE复活药也应及早使用。
3)要足量用药:
给药足量以保证快速和高效。
出现“阿托品化”:
瞳孔散大、口干、皮肤干燥、颜面潮红、肺部啰音显著减少或消失、心率加快等。
4)重复用药:
中、重度中毒或毒物不能从吸收部位彻底清除时,应重复给药,以巩固疗效。
AChE复活药:
氯解磷定
水溶液较稳定
明显减轻N样症状,对骨骼肌痉挛的抑制作用最为明显,能迅速抑制肌束颤动;对中枢神经系统的中毒症状也有一定的改善作用,但对M样症状的影响,应与阿托品合用,以控制症状。
碘解磷定的水溶液不稳定。
M胆碱受体阻断药:
阿托品:
药理作用和机制
1.腺体:
阿托品通过M胆碱受体的阻断作用抑制腺体分泌,对唾液腺和汗腺的作用最敏感。
泪腺和呼吸道腺体分泌也明显减少,较大剂量也减少胃液分泌。
2.眼:
出现扩瞳,眼内压升高和调节麻痹
3.平滑肌:
阿托品对多种内脏平滑肌有松弛作用,尤其对过度活动或痉挛的平滑肌作用更为显著。
可缓解胃肠绞痛,解除由药物引起的输尿管张力增高。
阿托品对胆管和子宫平滑肌作用较弱。
4.心脏
1)心率:
先慢后快,阿托品阻断了副交感神经节后纤维上的M1胆碱受体,从而减弱了突触中ACh对递质释放的负反馈作用所致。
较大剂量的阿托品通过阻断窦房结M2受体而解除了迷走神经对心脏的抑制作用,从而引起心率加快。
2)房室传导:
拮抗迷走神经兴奋过度所致的房室传导阻滞和心律失常。
阿托品尚可以缩短房室结的有效不应期,增加房颤或房扑患者的心室率。
5.血管与血压:
可以完全拮抗由胆碱酯类药物所引起的外周血管扩张和血压下降。
较大剂量的阿托品可引起皮肤血管扩张。
6.中枢神经系统:
较大剂量轻度兴奋中枢,中毒剂量可见明显中枢中毒症状,继续增加剂量,可见中枢兴奋转为抑制,最后可死于循环与呼吸衰竭。
试述阿托品的临床应用
答:
1.解除平滑肌痉挛:
各种内脏绞痛。
对胃肠绞痛,膀胱刺激症状如尿频、尿急等疗效较好,但对胆绞痛和肾绞痛疗效较差。
2.抑制腺体分泌:
用于全身麻醉前给药,以减少呼吸道腺体及唾液腺的分泌。
以及严重的盗汗和流涎症的治疗。
3.眼科:
1)虹膜睫状体炎
2)验光、眼底检查:
多用于儿童验光。
扩瞳检查眼底时持续时间较长。
4.缓慢型心律失常:
用于迷走神经过度兴奋所致的窦性心动过缓、窦房阻滞、房室传导阻滞等缓慢型心律失常。
5.抗休克:
大剂量用于感染性休克,能解除血管痉挛,舒张外周血管,改善微循环。
(休克伴有高热或心率过快者,不宜用阿托品。
)
6.解救有机磷酯类中毒。
(阿托品中毒可以用毒扁豆碱缓慢静脉注射,可迅速对抗阿托品中毒症状。
患者有明显的中枢兴奋时,可用地西泮对抗。
不可用吩噻嗪类药物,因此类药物具有M受体阻断作用而加重阿托品中毒症状。
)
禁忌症:
青光眼以及前列腺肥大者禁用阿托品。
东莨菪碱
治疗剂量出现中枢神经系统抑制。
抑制腺体分泌作用较阿托品强,扩瞳及调节麻痹作用较阿托品稍弱,对心血管系统作用较弱。
主要用于麻醉前给药,不仅抑制腺体分泌,而且具有中枢抑制作用。
可用于晕动病,与苯海拉明合用可增加疗效,以预防给药效果较好。
山莨菪碱(654)(人工合成的消旋品654-2)
抑制唾液分泌和扩瞳作用较弱,中枢兴奋作用较弱,对抗平滑肌痉挛和抑制心血管作用较弱。
对血管痉挛的解痉作用选择性相对较高。
主要用于感染性休克,也可用于内脏平滑肌绞痛。
毒性较低。
654-2与阿托品比较有哪些特点?
主要临床应用是什么?
答:
654-2的抑制唾液分泌和扩瞳作用较弱,仅为阿托品的1/20-1/10,不易进入中枢,中枢兴奋作用较弱,对抗平滑肌痉挛和抑制心血管作用较弱。
对血管痉挛的解痉作用选择性相对较高。
毒性较低。
主要临床应用是感染性休克,也可用于内脏平滑肌绞痛。
N胆碱受体阻断药:
琥珀胆碱:
(除极化型肌松药)
肌松作用快而短暂。
临床应用:
1.器官内插管、气管镜、食管镜检查等短时操作。
2.辅助麻醉(对清醒患者禁用)
不良反应:
窒息、眼内压升高、肌束颤动、血钾升高、心血管反应、恶性高热、增加腺体分泌、促进组胺释放。
在碱性溶液中可分解,不宜和硫喷妥钠混合使用。
凡可降低假性胆碱酯酶活性的药物都可以使其作用增加。
对琥珀胆碱发生的特异质反应是由于先天血浆胆碱酯酶缺乏所致。
筒箭毒碱(非除极化型肌松药)
临床可用作麻醉辅助药,用于胸腹手术和器官插管。
禁忌症为重症肌无力、支气管哮喘和严重休克。
简述琥珀胆碱和筒箭毒碱的区别。
答:
琥珀胆碱是除极化型肌松药。
其分子结构和ACh相似,与神经肌肉接头后膜的胆碱受体有较强亲和力,且在神经肌肉接头处不易被胆碱酯酶分解,因而产生与ACh相似但较持久的除极化作用。
造成对ACh的脱敏。
作用特点为:
1)最初可能出现短时肌束颤动。
2)连续用药可产生快速耐受性。
3)抗胆碱酯酶药不仅不能拮抗其肌松作用,又抑制了假性胆碱酯酶,使去极化肌松药不被水解,加重了病情。
4)治疗剂量并无神经节阻断作用。
筒箭毒碱是非除极化型肌松药。
能与ACh竞争神经肌肉接头的受体,但不激动受体,能竞争性阻断ACh的除极化作用,使骨骼肌松弛。
1)不产生肌束颤动,2)连续使用不产生耐受性。
3)过量可用适量的新斯的明解救4)有神经节阻断和释放组胺的作用,引起心率减慢、血压下降、支气管痉挛和唾液分泌增多等。
肾上腺素受体激动药:
Α肾上腺素受体激动药:
去甲肾上腺素:
(药用的NA是人工合成品,化学性质不稳定,见光、遇热易分解,在中性尤其是碱性溶液中迅速氧化变色而失效,在酸性溶液中较稳定,常用其重酒石酸盐。
)
NA进入机体迅速被摄取和代谢,故作用短暂,一般采用静脉滴注给药。
药理作用:
1.血管:
激动血管α1受体,使血管收缩,主要使小动脉和小静脉收缩。
(皮肤黏膜血管收缩最明显,其次是肾脏血管。
)动脉收缩使血流量减少,静脉的显著收缩使总外周阻力增加。
冠状血管舒张,主要是因为心脏兴奋,心肌的代谢产物(腺苷等)增加所致,同时因血压升高,提高冠状血管的灌注压,使冠脉流量增加。
激动α2受体,可反馈抑制NA的释放。
2.心脏:
较弱激动心脏的β1受体,使心肌收缩力加强,心率加快,传导加速,心排出量增加。
在整体情况下,心率由于血压
 升级会员
升级会员 