有参考基因组的转录组生物信息分析模板.docx
《有参考基因组的转录组生物信息分析模板.docx》由会员分享,可在线阅读,更多相关《有参考基因组的转录组生物信息分析模板.docx(28页珍藏版)》请在冰豆网上搜索。
有参考基因组的转录组生物信息分析模板
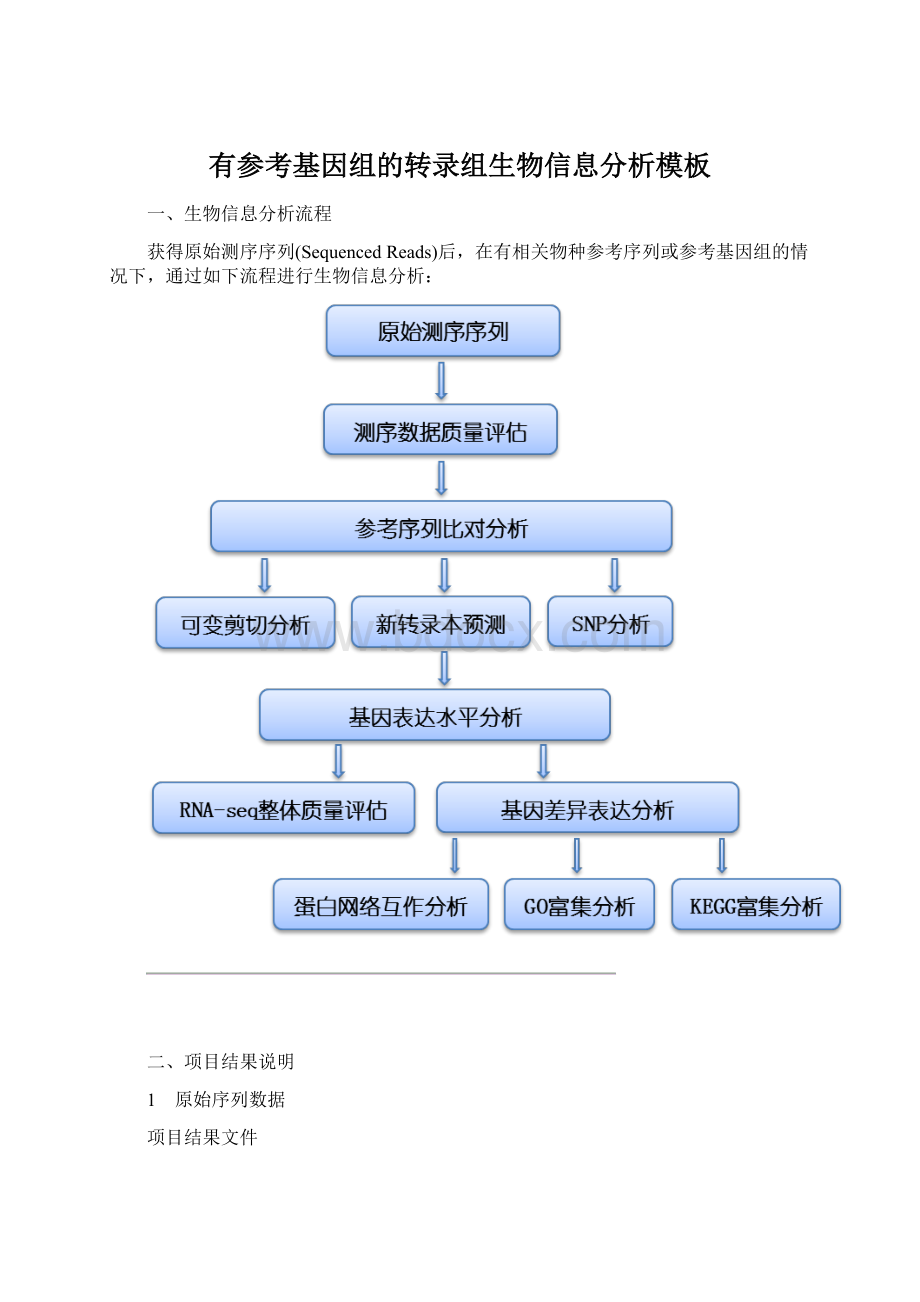
一、生物信息分析流程
获得原始测序序列(SequencedReads)后,在有相关物种参考序列或参考基因组的情况下,通过如下流程进行生物信息分析:
二、项目结果说明
1 原始序列数据
项目结果文件
高通量测序(如illuminaHiSeqTM2000/MiSeq等测序平台)测序得到的原始图像数据文件经碱基识别(BaseCalling)分析转化为原始测序序列(SequencedReads),我们称之为RawData或RawReads,结果以FASTQ(简称为fq)文件格式存储,其中包含测序序列(reads)的序列信息以及其对应的测序质量信息。
测序样品真实数据随机截取展示
FASTQ格式文件中每个read由四行描述,如下:
@EAS139:
136:
FC706VJ:
2:
2104:
15343:
1973931:
Y:
18:
ATCACG
GCTCTTTGCCCTTCTCGTCGAAAATTGTCTCCTCATTCGAAACTTCTCTGT
+
@@CFFFDEHHHHFIJJJ@FHGIIIEHIIJBHHHIJJEGIIJJIGHIGHCCF
其中第一行以“@”开头,随后为illumina测序标识符(SequenceIdentifiers)和描述文字(选择性部分);第二行是碱基序列;第三行以“+”开头,随后为illumina测序标识符(选择性部分);第四行是对应序列的测序质量(Cocketal.)。
illumina测序标识符详细信息如下:
EAS139
Uniqueinstrumentname
136
RunID
FC706VJ
FlowcellID
2
Flowcelllane
2104
Tilenumberwithintheflowcelllane
15343
'x'-coordinateoftheclusterwithinthetile
197393
'y'-coordinateoftheclusterwithinthetile
1
Memberofapair,1or2(paired-endormate-pairreadsonly)
Y
Yifthereadfailsfilter(readisbad),Notherwise
18
0whennoneofthecontrolbitsareon,otherwiseitisanevennumber
ATCACG
Indexsequence
第四行中每个字符对应的ASCII值减去33,即为对应第二行碱基的测序质量值。
如果测序错误率用e表示,illuminaHiSeqTM2000/MiSeq的碱基质量值用Qphred表示,则有下列关系:
公式一:
Qphred = -10log10(e)
illuminaCasava1.8版本测序错误率与测序质量值简明对应关系如下:
测序错误率
测序质量值
对应字符
5%
13
.
1%
20
5
0.1%
30
?
0.01%
40
I
2 测序数据质量评估
项目结果文件
2.1 测序错误率分布检查
每个碱基测序错误率是通过测序Phred数值(Phredscore,Qphred)通过公式1转化得到,而Phred数值是在碱基识别(BaseCalling)过程中通过一种预测碱基判别发生错误概率模型计算得到的,对应关系如下表所显示:
illuminaCasava1.8版本碱基识别与Phred分值之间的简明对应关系
Phred分值
不正确的碱基识别
碱基正确识别率
Q-sorce
10
1/10
90%
Q10
20
1/100
99%
Q20
30
1/1000
99.9%
Q30
40
1/10000
99.99%
Q40
测序错误率与碱基质量有关,受测序仪本身、测序试剂、样品等多个因素共同影响。
对于RNA-seq技术,测序错误率分布具有两个特点:
(1)测序错误率会随着测序序列(SequencedReads)的长度的增加而升高,这是由于测序过程中化学试剂的消耗而导致的,并且为illumina高通量测序平台都具有的特征(ErlichandMitra,2008;Jiangetal.)。
(2)前6个碱基的位置也会发生较高的测序错误率,而这个长度也正好等于在RNA-seq建库过程中反转录所需要的随机引物的长度。
所以推测前6个碱基测序错误率较高的原因为随机引物和RNA模版的不完全结合(Jiangetal.)。
测序错误率分布检查用于检测在测序长度范围内,有无异常的碱基位置存在高错误率,比如中间位置的碱基测序错误率显著高于其他位置。
一般情况下,每个碱基位置的测序错误率都应该低于0.5%。
图2.1 测序错误率分布图
横坐标为reads的碱基位置,纵坐标为单碱基错误率
2.2 GC含量分布检查
GC含量分布检查用于检测有无AT、GC分离现象,而这种现象可能是测序或者建库所带来的,并且会影响后续的定量分析。
在illumina测序平台的转录组测序中,反转录成cDNA时所用的6bp的随机引物会引起前几个位置的核苷酸组成存在一定的偏好性。
而这种偏好性与测序的物种和实验室环境无关,但会影响转录组测序的均一化程度(Hansenetal.)。
除此之外,理论上G和C碱基及A和T碱基含量每个测序循环上应分别相等,且整个测序过程稳定不变,呈水平线。
对于DGE测序来说,由于随机引物扩增偏差等原因,常常会导致在测序得到的每个read前6-7个碱基有较大的波动,这种波动属于正常情况。
图2.2 GC含量分布图
横坐标为reads的碱基位置,纵坐标为单碱基所占的比例;不同颜色代表不同的碱基类型
2.3 测序数据过滤
测序得到的原始测序序列,里面含有带接头的、低质量的reads,为了保证信息分析质量,必须对rawreads进行过滤,得到cleanreads,后续分析都基于cleanreads。
数据处理的步骤如下:
(1)去除带接头(adapter)的reads;
(2)去除N(N表示无法确定碱基信息)的比例大于10%的reads;
(3)去除低质量reads。
RNA-seq的接头(Adapter,OligonucleotidesequencesforTruSeqTMRNAandDNASamplePrepKits)信息:
RNA5’Adapter(RA5),part#15013205:
5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3’
RNA3’Adapter(RA3),part#15013207:
5’-GATCGGAAGAGCACACGTCTGAACTCCAGTCAC(6位index)ATCTCGTATGCCGTCTTCTGCTTG-3’
图2.3 原始数据过滤结果
2.4 测序数据质量情况汇总
表2.4 数据产出质量情况一览表
Samplename
Rawreads
Cleanreads
cleanbases
Errorrate(%)
Q20(%)
Q30(%)
GCcontent(%)
HS1_1
36579608
35175205
3.52G
0.03
97.88
92.88
49.39
HS1_2
36579608
35175205
3.52G
0.03
96.50
90.38
49.59
HS2_1
36547734
35119463
3.51G
0.03
97.85
92.81
49.53
数据质量情况详细内容如下:
(1)Rawreads:
统计原始序列数据,以四行为一个单位,统计每个文件的测序序列的个数。
(2)Cleanreads:
计算方法同RawReads,只是统计的文件为过滤后的测序数据。
后续的生物信息分析都是基于Cleanreads。
(3)Cleanbases:
测序序列的个数乘以测序序列的长度,并转化为以G为单位。
(4)Errorrate:
通过公式1计算得到。
(5)Q20、Q30:
分别计算Phred数值大于20、30的碱基占总体碱基的百分比。
(6)GCcontent:
计算碱基G和C的数量总和占总的碱基数量的百分比。
3 参考序列比对分析
项目结果文件
测序序列定位算法:
根据不同的基因组的特征,我们选取相对合适的软件(动植物用TopHat(Trapnelletal.,2009)、真菌或者基因密度较高的物种用Bowtie),合适的参数设置(如最大的内含子长度,会根据已知的该物种的基因模型来进行统计分析),将过滤后的测序序列进行基因组定位分析。
下图为TopHat的算法示意图:
Tophat的算法主要分为两个部分:
(1)将测序序列整段比对到外显子上。
(2)将测序序列分段比对到两个外显子上。
我们统计了实验所产生的测序序列的定位个数(TotalMappedReads)及其占cleanreads的百分比,其中包括多个定位的测序序列个数(MultipleMappedReads)及其占总体(cleanreads)的百分比,以及单个定位的测序序列个数(UniquelyMappedReads)及其占总体(cleanreads)的百分比。
3.1 Reads与参考基因组比对情况统计
表3.1 Reads与参考基因组比对情况一览表
Samplename
HS1
HS2
HT1
HT2
HW1
HW2
Totalreads
70350410
70238926
76161678
50666084
46573662
40543118
Totalmapped
60529821(86.04%)
60232484(85.75%)
63555439(83.45%)
43461327(85.78%)
40246848(86.42%)
34971284(86.26%)
Multiplemapped
606556(0.86%)
633575(0.9%)
714678(0.94%)
450156(0.89%)
389470(0.84%)
335509(0.83%)
Uniquelymapped
59923265(85.18%)
59598909(84.85%)
62840761(82.51%)
43011171(84.89%)
39857378(85.58%)
34635775(85.37%)
Read-1
30176973(42.9%)
29987004(42.69%)
31592931(41.48%)
21654629(42.74%)
20028779(43%)
17411209(43.02%)
Read-2
29746292(42.28%)
29611905(42.16%)
31247830(41.03%)
21356542(42.15%)
19828599(42.57%)
17224566(42.35%)
Readsmapto'+'
29930036(42.54%)
29783311(42.4%)
31409912(41.24%)
21476601(42.39%)
19923501(42.78%)
17289330(42.61%)
Readsmapto'-'
29993229(42.63%)
29815598(42.45%)
31430849(41.27%)
21534570(42.5%)
19933877(42.8%)
17346445(42.76%)
Non-splicereads
42357242(60.21%)
42528691(60.55%)
45227757(59.38%)
31347392(61.87%)
28062847(60.25%)
24725216(61.1%)
Splicereads
17566023(24.97%)
17070218(24.3%)
17613004(23.13%)
11663779(23.02%)
11794531(25.32%)
9910559(24.26%)
Readsmappedinproperpairs
53795182(76.47%)
54428240(77.49%)
56181352(73.77%)
38524314(76.04%)
36101400(77.51%)
31246362(77.25%)
比对结果统计详细内容如下:
(1)Totalreads:
测序序列经过测序数据过滤后的数量统计(Cleandata)。
(2)Totalmapped:
能定位到基因组上的测序序列的数量的统计;一般情况下,如果不存在污染并且参考基因组选择合适的情况下,这部分数据的百分比大于70%。
(3)Multiplemapped:
在参考序列上有多个比对位置的测序序列的数量统计;这部分数据的百分比一般会小于10%。
(4)Uniquelymapped:
在参考序列上有唯一比对位置的测序序列的数量统计。
(5)Readsmapto'+',Readsmapto'-':
测序序列比对到基因组上正链和负链的统计。
(6)Splicereads:
(2)中,分段比对到两个外显子上的测序序列(也称为Junctionreads)的统计,Non-splicereads为整段比对到外显子的将测序序列的统计,Splicereads的百分比取决于测序片段的长度。
3.2 Reads在参考基因组不同区域的分布情况
对Totalmappedreads的比对到基因组上的各个部分的情况进行统计,定位区域分为Exon(外显子)、Intron(内含子)和Intergenic(基因间隔区域)。
正常情况下,Exon(外显子)区域的测序序列定位的百分比含量应该最高,定位到Intron(内含子)区域的测序序列可能是由于非成熟的mRNA的污染或者基因组注释不完全导致的,而定位到Intergenic(基因间隔区域)的测序序列可能是因为基因组注释不完全以及背景噪音。
图3.2 Reads在参考基因组不同区域的分布情况
3.3 Reads在染色体上的密度分布情况
对Totalmappedreads的比对到基因组上的各个染色体(分正负链)的密度进行统计,如下图所示,具体作图的方法为用滑动窗口(windowsize)为1K,计算窗口内部比对到碱基位置上的reads的中位数,并转化成log2。
正常情况下,整个染色体长度越长,该染色体内部定位的reads总数会越多(Marquezetal.)。
从定位到染色体上的reads数与染色体长度的关系图中,可以更加直观看出染色体长度和reads总数的关系。
图3.3 Reads在染色体上的密度分布图
上图:
横坐标为染色体的长度信息(以百万碱基为单位),纵坐标为log2(reads的密度的中位数),绿色为正链,红色为负链下图:
横坐标为染色体的长度信息(单位为Mb),纵坐标为mapped到染色体上的reads数(单位为M)
3.4 Reads比对结果可视化
我们提供RNA-seqReads在基因组上比对结果的bam格式文件,部分物种还提供相应的参考基因组和注释文件,并推荐使用IGV(IntegrativeGenomicsViewer)浏览器对bam文件进行可视化浏览。
IGV浏览器具有以下特点:
(1)能在不同尺度下显示单个或多个读段在基因组上的位置,包括读段在各个染色体上的分布情况和在注释的外显子、内含子、剪接接合区、基因间区的分布情况等;
(2)能在不同尺度下显示不同区域的读段丰度,以反映不同区域的转录水平;(3)能显示基因及其剪接异构体的注释信息;(4)能显示其他注释信息;(5)既可以从远程服务器端下载各种注释信息,又可以从本地加载注释信息。
IGV浏览器使用方法可参考我们提供的使用说明文档(IGVQuickStart.pdf)。
图3.4 IGV浏览器界面
4 可变剪切分析
项目结果文件
用ASprofile软件对Cufflinks(Trapnelletal.)预测出的基因模对每个样品的可变剪切事件分别进行分类和表达量统计。
分析流程及ASprofile中的可变剪切事件分类如下图所示:
12类可变剪切事件定义如下:
(1)TSS:
Alternative5'firstexon(transcriptionstartsite)第一个外显子可变剪切
(2)TTS:
Alternative3'lastexon(transcriptionterminalsite) 最后一个外显子可变剪切
(3)SKIP:
Skippedexon(SKIP_ON,SKIP_OFFpair) 单外显子跳跃
(4)XSKIP:
ApproximateSKIP(XSKIP_ON,XSKIP_OFFpair) 单外显子跳跃(模糊边界)
(5)MSKIP:
Multi-exonSKIP(MSKIP_ON,MSKIP_OFFpair) 多外显子跳跃
(6)XMSKIP:
ApproximateMSKIP(XMSKIP_ON,XMSKIP_OFFpair) 多外显子跳跃(模糊边界)
(7)IR:
Intronretention(IR_ON,IR_OFFpair) 单内含子滞留
(8)XIR:
ApproximateIR(XIR_ON,XIR_OFFpair) 单内含子滞留(模糊边界)
(9)MIR:
Multi-IR(MIR_ON,MIR_OFFpair) 多内含子滞留
(10)XMIR:
ApproximateMIR(XMIR_ON,XMIR_OFFpair) 多内含子滞留(模糊边界)
(11)AE:
Alternativeexonends(5',3',orboth) 可变5'或3'端剪切
(12)XAE:
ApproximateAE 可变5'或3'端剪切(模糊边界)
4.1 可变剪切事件分类和数量统计
图4.1 AS分类和数量统计
纵轴为可变剪切事件的分类缩写,横轴为该种事件下可变剪切的数量,不同样品用不同子图和颜色区分
4.2 可变剪切事件结构和表达量统计
表4.2 AS结构和表达量统计
event_id
event_type
gene_id
chrom
event_start
event_end
event_pattern
strand
fpkm
ref_id
1000001
TSS
CUFF.100
1
3438277
3438330
3438330
+
1.0000000000
ENSGALT00000010225
1000002
TSS
CUFF.100
1
3450218
3450253
3450253
+
3.0000000000
ENSGALT00000010225
1000003
TSS
CUFF.100
1
3456744
3457165
3457165
+
2.0000000000
ENSGALT00000010225
1000004
TTS
CUFF.100
1
3474806
3478178
3474806
+
5.0000000000
ENSGALT00000010225
(1)event_id:
AS事件编号
(2)event_type:
AS事件类型(TSS,TTS,SKIP_{ON,OFF},XSKIP_{ON,OFF},MSKIP_{ON,OFF},XMSKIP_{ON,OFF},IR_{ON,OFF},XIR_{ON,OFF},AE,XAE)
(3)gene_id:
cufflink组装结果中的基因编号
(4)chrom:
染色体编号
(5)event_start:
AS事件起始位置
(6)event_end:
AS事件结束位置
(7)event_signature:
AS事件特征(forTSS,TTS-insideboundaryofalternativemarginalexon;for*SKIP_ON,thecoordinatesoftheskippedexon(s);for*SKIP_OFF,thecoordinatesoftheenclosingintrons;for*IR_ON,theendcoordinatesofthelong,intron-containingexon;for*IR_OFF,thelistingofcoordinatesofalltheexonsalongthepathcontainingtheretainedintron;for*AE,thecoordinatesoftheexonvariant)
(8)strand:
基因正负链信息
(9)fpkm:
此AS类型该基因表达量
(10)ref_id:
此基因在参考注释文件中的编号
5 新转录本预测
项目结果文件
将所有测序reads数据的基因组定位结果放到一起,用Cufflinks进行组装,然后用Cuffcompare和已知的基因模型进行比较,可以:
(1)发现新的未知基因(相对于原有基因注释文件);
(2)发现已知基因新的外显子区域;(3)对已知基因的起始和终止位置进行优化。
新基因和新外显子区域预测结果为GTF格式的注释文件。
GTF格式的详细说明可参考(http:
//mblab.wustl.edu/GTF22.html)
表5.1 新转录本结构注释结果
seqname
source
feature
start
end
score
strand
frame
attributes
1
novelGene
exon
18531
19499
.
+
.
gene_id"Novel00001";transcript_id"Novel00001.1";exon_number"1";
1
novelGene
exon
20813
21813
.
+
.
gene_id"Novel00002";transcript_id"Novel00002.1";exon_number"1";
1
novelGene
exon
23917
24402
.
+
.
gene_id"Novel00003";transcript_id"Novel000
 升级会员
升级会员 