Softberry网站部分功能介绍_精品文档.doc
《Softberry网站部分功能介绍_精品文档.doc》由会员分享,可在线阅读,更多相关《Softberry网站部分功能介绍_精品文档.doc(8页珍藏版)》请在冰豆网上搜索。
Softberry网站部分使用功能介绍
摘要
Softberry是专业的在线比对寻找生物启动子和启动子模型的网站,其中有很多功能都是我们在以后的生物信息学研究中将要使用到的,所以我们有必要了解其使用方法,本文主要介绍了它的功能的两个部分,启动子序列比对功能和SELTAG软件的使用简介。
关键词:
启动子预测启动子模型SELTAG软件
ABSTRACK
Softberryisaprofessionalonlineprogramwhichlookingforbiologicalpromoterandthemodelofpromoter,therearealotoffeaturesusageonthiswebsitewillbehelpedinourbioinformaticsresearch,soitisnecessarytoknowitsusingmethod,thispapermainlyintroducesitsfunctionoftwoparts,thepromotersequencealignmentfunctionandtheusageofSELTAGsoftwareprofile.
Keywords:
promoterpredictionpromotermodelSELTAGsoftware
第一部分:
Searchforpromoters/functionalmotifs
寻找启动子/功能性序列
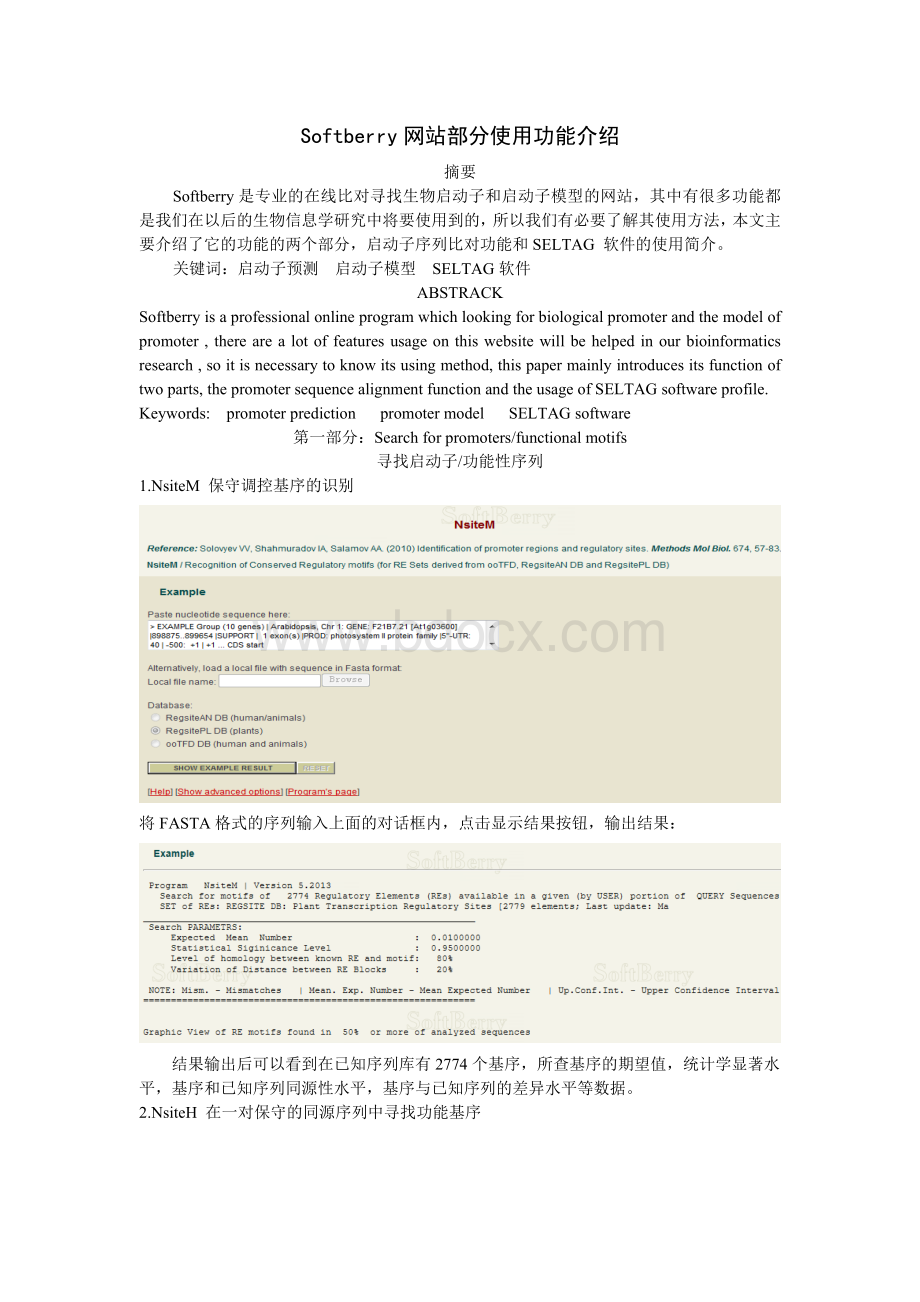
1.NsiteM保守调控基序的识别
将FASTA格式的序列输入上面的对话框内,点击显示结果按钮,输出结果:
结果输出后可以看到在已知序列库有2774个基序,所查基序的期望值,统计学显著水平,基序和已知序列同源性水平,基序与已知序列的差异水平等数据。
2.NsiteH在一对保守的同源序列中寻找功能基序
在对话框内分别输入两条要比较的序列,然后点击下面的显示结果按钮,输出结果:
在结果内可以看到两条序列比对的结果,期望值,统计学显著水平,最小保守水平,同源水平,变异水平的数据。
3.POLYAH识别3’末端多聚核苷酸尾区域
将FASTA格式的序列输入上面的对话框里,按输出结果按钮,可以得到以下数据:
输出结果表明所查序列中有两个ployA尾位点被预测到。
4.BPROM细菌启动子的预测
将细菌的所查序列粘贴到此处,点击显示结果按钮,输出结果:
结果显示,我们所查序列的名称和总长度,以及预测出来的启动子所在位置和其对应的分值。
5.PromH(G)在真核生物中利用同源序列进行启动子预测
首先,将一段序列与另一段同源序列分别输入以上两个对话框内,然后点击数据出结果按钮:
可得如下结果:
PHa表示比对序列在局部搜索区域的同源性水平:
PHs表示比对序列在TSS系列软件中的同源性水平;PHss表示比对序列直接在TSS软件中的同源性水平;PHt表示TATA-BOX在比对序列中的同源性水平;PHr代表调控因子在局部搜索区域的同源性水平。
下面的就是各项的比对结果与其分值。
6.PromH(W)在真核基因组使用同源序列预测启动子(仅供学术使用)
先将两条要比对的序列输入上边的两个对话框,点击输出结果按钮可得:
与PromH(G)不同,这里的输出结果多出两条:
Initial/FinalThresholdsforTATA+promoters-0.10/2.50
TATA+启动子的最初的/最终的阈值-0.10/2.50
Initial/FinalThresholdsforTATA-/enhancers-0.70/3.70
TATA-/增强子的最初/最终阈值-0.70/3.70
7.CgGFinderCpG寻找器,序列中GC区域的寻找
将要寻找GC区域的序列输入上面的对话框内,可得下图:
程序找到4个GC区域,分别排列在下方,它们的各项指标都在下面显示,起始位点和终止位点和长度等。
8.ScanWM-p寻找植物调控序列的权重矩阵模型
将要寻找模型的序列输入此处,点击显示结果,可得:
这样,在相应数据库中找到的模型就会排布在下方,
总共有5个模型被找到。
第二部分:
SELTAG软件简介
seltag是用于分析基因表达数据的最完美的工具。
它具有以下功能:
1.它可以分析所有或选定的基因组或组织;
2.在复杂选择标准的基础上,对组织特异性基因进行选择;
3.对数据进行可视化表达;
4.用来识别在一系列组织中表达相同(相关)的基因;
5.选择特定的基因,如与某些疾病有关的受体或分泌蛋白。
seltag允许用户:
① 通过基因的表达水平或其他在不同的实验中获得的参数来选择基因;
② 通过基因在不同实验中的表达水平来确定基因的排序;
③ 用先进的绘图工具将基因表达谱可视化;
④ 利用相似的表达谱寻找一个或多个基因;
⑤ 评估两个或多个基因的表达谱之间的相关性;
⑥ 通过基因表达水平相似的基因表达谱与实验相似的聚类基因表达数据进行基因或组织层次聚类和显示结果相似的树图;
⑦ 分析使用主成分分析法的相关基因表达谱的协方差矩阵和可视化的结果;
⑧ 它还可以结合外部数据库分析检索的数据(例如UniGene数据库)
上图就是可视化基因表达水平的一个软件截图,对比的是人肿瘤细胞与人正常细胞某些共有基因的表达情况。
可以清晰地反应出肿瘤细胞中哪些基因的表达量明显提高。
 升级会员
升级会员 