色酮3甲酸的合成工艺研究.docx
《色酮3甲酸的合成工艺研究.docx》由会员分享,可在线阅读,更多相关《色酮3甲酸的合成工艺研究.docx(11页珍藏版)》请在冰豆网上搜索。
色酮3甲酸的合成工艺研究
色酮-3-甲酸的合成工艺研究
作者:
仲芯颖褚朝森李天雪王晓丽胡玉涛
来源:
《当代化工》2020年第11期
摘要:
以色酮-3-甲醛为原料制备色酮-3-甲腈,后经氢氧化钾水解制备色酮-3-甲酸。
考察不同溶剂、氢氧化钾用量、氢氧化钾浓度、温度、时间对水解反应的影响。
结果表明,最佳反应条件为:
乙二醇做溶剂、氢氧化钾与色酮-3-甲腈物质的量比为5∶1、氢氧化钾浓度为9.7mol·L-1、反应温度120℃、反应时间5h,在上述条件下水解反应收率为90%。
两步反应总收率为70%。
产物经高效液相色谱检测,纯度为98.17%。
关键词:
色酮-3-甲酸;色酮-3-甲醛;色酮-3-甲腈;水解
中图分类号:
TQ031.5文献标识码:
A文章编号:
1671-0460(2020)11-2423-04
ResearchonSynthesisofChromone3-CarboxylicAcid
ZHONGXin-ying1,CHUChao-sen1,2,LITian-xue1,2,WANGXiao-li1,HUYu-Tao1
(1.LianyungangTCMBranchofJiangsuUnionTechnicalInstitute,Lianyungang222007,China;
2.LianyungangCommonTechnologicalCenterforDrugResearchandDevelopment,Lianyungang222007,China)
Abstract:
Usingchromone-3-carboxaldehydeasstartingmaterial,3-cyanochromonewasprepared.Chromone3-carboxylicacidwassynthesizedviahydrolysisof3-cyanochromonebypotassiumhydroxide.Theeffectofsolvent,potassiumhydroxidedosage,potassiumhydroxideconcentration,temperature,timeonhydrolysisreactionwasstudied.Theresultshowedthattheoptimumreactionconditionwasasfollows:
usingethyleneglycolassolvent,moleratioofpotassiumhydroxideand3-cyanochromone5∶1,potassiumhydroxideconcentration9.7mol·L-1,120℃,reactiontime5h.Underaboveconditions,theyieldofhydrolysisreactionwas90%.Thetotalyieldofthetwo-stepreactionwas70%.TheproductpuritydeterminedbyHPLCwas98.17%.
Keywords:
3-Carboxylicacid;Chromone-3-carboxaldehyde;3-Cyanochromone;Hydrolysis
色酮-3-甲酸(chromone3-carboxylicacid)是色酮的衍生物[1],廣泛存在于自然界中,具有抗炎、抗菌、抗癌等生物活性[2-3]。
其分子中含有羧基,可以制成酯、酰胺、酰氯等[4-5]有价值的有机合成中间体物质。
目前色酮-3-甲酸的合成方法已有报道。
曹玲华[6]等采用Jones氧化法,以色酮-3-甲醛为原料,在硫酸存在下,以丙酮为溶剂,用三氧化铬氧化制得产物,收率39%。
该方法操作简便,但收率低,不经济。
TARUN[7]等报道采用色酮-3-甲醛与硝基甲烷在锌和冰乙酸存在下制备硝酮化合物,后用硫酸在110~115℃下水解制备产物,收率90%。
该方法中使用的原料硝基甲烷为危险化学品,不利于工业化生产。
AMBARTSUMYAN[8]等用色酮-3-甲醛在六甲基磷酰三胺和苯存在下制备产物,收率5%,该方法收率太低,不可取。
YOSHIMASA[9]等报道用色酮-3-甲醛与N-溴代丁二酰亚胺溴代后经水解制备产物,收率达93%。
该方法需要用四氯化碳为溶剂在光照条件下完成,且溶剂使用量较大,难以放大生产。
总体看来,目前色酮-3-甲酸的合成方法存在着收率低、操作难度大、原料危险性高等问题。
REDDY[10]等报道色酮-3-甲醛在盐酸羟胺、碘化钠存在下,乙腈为溶剂,回流反应可以制得色酮-3-甲腈,成功在色酮母核的3号位引入氰基。
氰基可发生水解、还原等多种化学反应转化为有价值的中间体化合物[11]。
HUANG[12]等报道了用氢氧化锂水解氰基制备羧酸的方法,但氢氧化锂危险性高,价格昂贵,不宜采用。
本研究以此为切入点,设计采用色酮-3-甲醛为原料,与盐酸羟胺、碘化钠在乙腈溶剂中回流反应制备色酮-3-甲腈,后经氢氧化钾水解制备目标产物,反应过程如图1所示。
1实验部分
1.1实验材料
色酮-3-甲醛,分析纯(质量分数99%),上海阿拉丁生化科技股份有限公司;乙二醇,工业级(质量分数95%),上海沃化化工有限公司;其余试剂均为市售分析纯试剂。
1HNMR在德国Bruker公司AM500MHz共振仪上测定;熔点在上海精密科学仪器有限公司WRS-B型熔点仪上测定;纯度在日本岛津高效液相色谱仪(LC-2010型)上测定。
1.2实验方法
1.2.1色酮-3-甲腈的制备
参照文献[10]方法,取1L三颈瓶,加入色酮-3-甲醛110.4g(0.63mol),盐酸羟胺52.8g(0.76mol),碘化钠47.52g(0.26mol),加入乙腈540mL,加熱回流10h,TLC监测反应完全。
反应液冷却至室温,过滤,100mL乙腈洗涤滤饼,收集滤液,减压浓缩除去溶剂得灰黑色固体。
固体用2L二氯甲烷溶解,溶液依次用2L质量分数为5%地亚硫酸钠溶液洗涤一次,2L纯化水洗涤2次,有机相用无水硫酸钠干燥2h,减压浓缩除去溶剂得灰黑色固体。
固体置于1L三颈瓶中,加入500mL甲醇,搅拌下加热回流30min,移除热源,继续搅拌冷却至室温,过滤得灰色固体产品83.5g(0.49mol),该步反应收率78%。
熔点:
174~175℃;MS:
171.1(M+);1HNMR(DMSO):
δ=7.41-7.58(m,2H,ArH);δ=7.78(m,1H,ArH);δ=8.1(dd,1H,ArH);δ=8.8(s,1H,CH=)。
1.2.2色酮-3-甲酸的制备
取色酮-3-甲腈50g(0.29mol)于500mL三颈瓶中,加入乙二醇150mL,搅拌下缓慢加入氢氧化钾81g(1.45mol),此时有明显放热现象,产生大量气泡,待气泡消失,加热反应液至120℃,维持温度8h。
移除热源,待反应液降温至室温,向反应液中加入纯化水150mL,搅拌均匀后将反应液移至1L的分液漏斗中,用二氯甲烷300mL萃取两次,水相倒入500mL烧杯中,冰水浴下缓慢滴加浓盐酸调节至体系pH值为3,继续搅拌2h,析出大量黄色固体,过滤,收集固体。
固体用甲醇重结晶后得终产物色酮-3-甲酸49g(0.26mol),该步反应收率90%。
两步反应总收率70%;熔点:
201~203℃;MS:
190.1(M+);1HNMR(DMSO):
δ=7.62(t,1H,ArH);δ=7.81(t,1H,ArH);δ=7.94(m,1H,ArH);δ=8.17(m,1H,ArH);δ=9.13(s,1H,CH=);δ=13.25(s,1H,OH)。
1.3高效液相色谱(HPLC)检测产物纯度
色谱柱:
岛津Shim-packVP-ODS型色谱柱(150mm×4.6mm,5μm)。
流动相:
甲醇∶水(含质量分数0.1%甲酸)=55∶45。
检测波长:
216nm;柱温:
30℃;进样量:
10μL;流速:
1.0mL·min-1。
2结果与讨论
2.1色酮-3-甲腈的合成
参考文献[10]方法进行色酮-3-甲腈的合成,投料量是文献的6.3倍,研究表明,原料用量增加,反应时间延长,需要10h,明显长于文献报道时间。
溶解粗产物需要的溶剂二氯甲烷的量显著增加,实验过程处理难度增大,最终收率78%,略低于文献报道数据。
2.2色酮-3-甲酸的合成
文献[13]报道色酮-3-甲腈在浓盐酸中水解得到产物色酮-3-甲酸,收率较低,为55%。
为提高收率,本研究采用氢氧化钾在碱性条件下进行水解,参考文献[14-15]方法,实验考察了不同溶剂、氢氧化钾用量、氢氧化钾浓度、温度、时间对反应的影响。
2.2.1不同溶剂的影响
采用色酮-3-甲腈0.29mol,氢氧化钾0.58mol,溶剂量200mL,反应2h,考察不同溶剂对水解反应的影响,结果见表1。
实验结果表明,不同溶剂中水解反应结果不同。
其中,水为溶剂时,在回流条件下收率最低,仅8%,这是由于色酮-3-甲腈难溶于水,不能和氢氧化钾分子有效接触,水解反应难以进行;甲醇和乙醇为溶剂时水解反应收率接近,其中乙醇略高于甲醇,这是由于乙醇的沸点较高,回流状态下优于甲醇;乙二醇在100℃下水解收率最高,达45%,由于乙二醇具有较高的沸点(196~198℃),温度可变范围大,是本反应最佳的溶剂。
2.2.2氢氧化钾用量的影响
采用色酮-3-甲腈0.29mol,乙二醇200mL,100℃,反应2h,参考文献[16]方法,实验考察氢氧化钾不同用量对水解反应的影响,结果见表2。
实验结果表明,随着氢氧化钾用量的增加,水解反应收率逐渐增加,当氢氧化钾与色酮-3-甲腈摩尔比为5∶1时,收率达58%,之后继续增加氢氧化钾用量,收率变化不大,综合考虑,5∶1为最佳用量。
2.2.3氢氧化钾浓度的影响
采用色酮-3-甲腈0.29mol,氢氧化钾1.45mol,100℃,反应2h,考察氢氧化钾不同浓度对水解反应的影响,结果见表3。
实验结果表明,溶剂用量少,氢氧化钾浓度高有利于水解反应进行,序号2、3表现出较高的收率,最高达69%;当浓度过高时(序号1),溶质难以均匀分散,反应液较黏稠,受热不均匀,导致反应结果较差,收率仅48%;当浓度较低时(序号4、5、6),分子间接触机会变小,反应结果不理想,收率均低于60%。
研究证实,氢氧化钾浓度为9.7mol·L-1(乙二醇用量为150mL)时收率最高,为69%,故确定9.7mol·L-1为最佳氢氧化钾浓度。
2.2.4反应温度的影响
采用色酮-3-甲腈0.29mol,氢氧化钾1.45mol,乙二醇150mL,反应2h,考察不同温度对水解反应的影响,结果见表4。
实验结果表明,随着反应温度的升高,水解反应收率呈上升趋势,证明高温有利于反应的发生。
当温度升至120℃时,收率达78%,之后随着温度的升高,收率趋于平缓,变化不大。
从节约能源的角度考虑,选择120℃为最佳反应温度。
2.2.5反应时间的影响
采用色酮-3-甲腈0.29mol,氢氧化钾1.45mol,乙二醇150mL,120℃,考察不同时间对水解反应的影响,结果见表5。
实验结果表明,随着反应时间的延长,收率呈上升趋势,当反应时间为5h,收率达90%,之后延长反应时间,收率提升不明显,故确定5h为最佳反应时间。
综上所述,色酮-3-甲腈水解制备色酮-3-甲酸的最佳反应条件为乙二醇做溶剂、氢氧化钾与色酮-3-甲腈物质的量比为5∶1、氢氧化钾浓度为9.7mol·L-1、反应温度120℃、反应时间5h,此时反应收率最高,达90%。
2.3产物纯度的检测
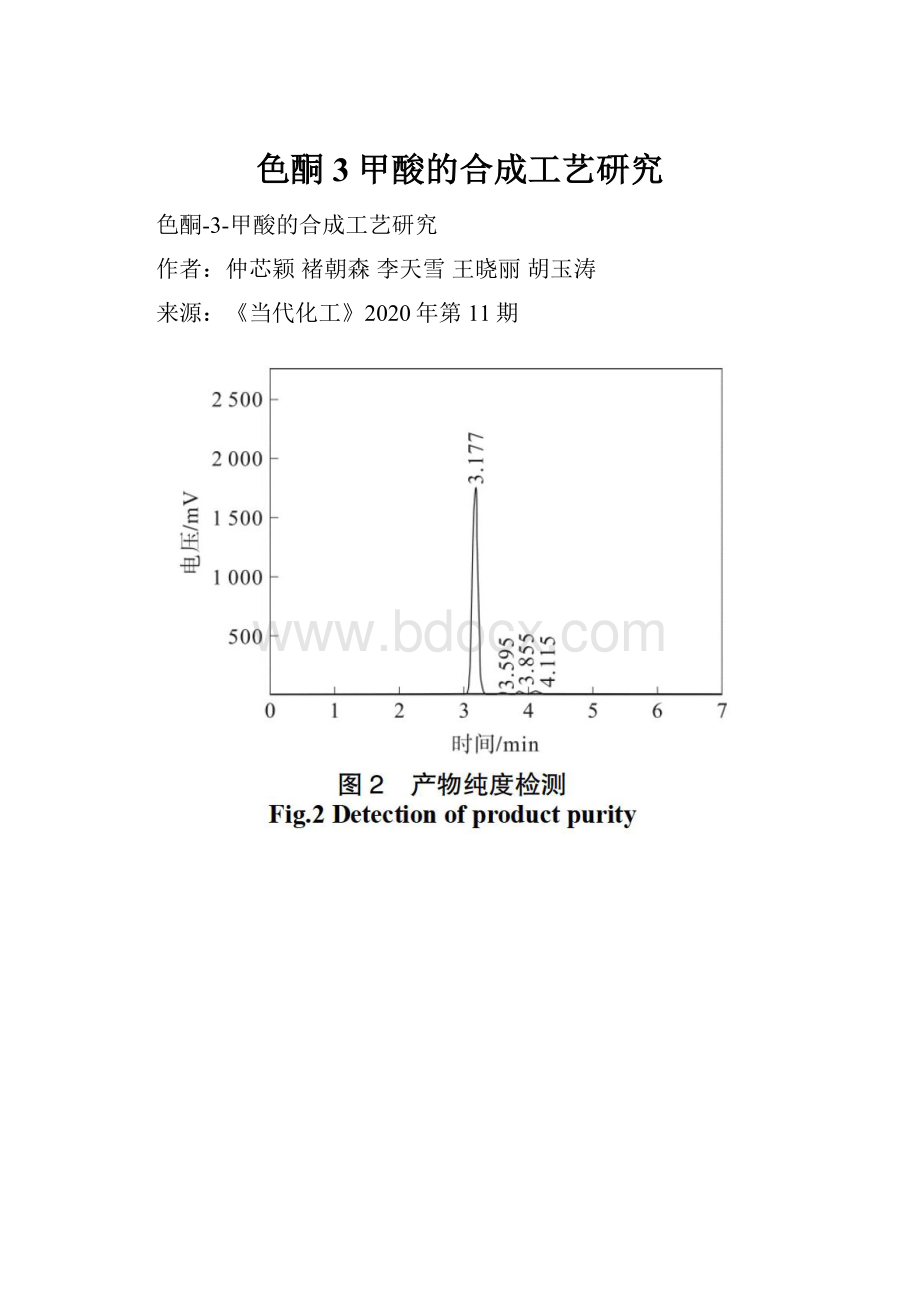
将待测样品配制成0.5mg·mL-1的甲醇溶液,在216nm波长下,采用甲醇∶水(含质量分数0.1%甲酸)体积比55∶45的溶液为流动相,控制柱温30℃,流速1.0mL·min-1,檢测产物纯度,结果见图2。
结果表明,产物出峰时间为3.177min,杂质出峰时间分别为3.595、3.855、4.115min,产物纯度98.17%。
色酮-3-甲酸在甲醇和水中溶解性不佳,流动相中加入质量分数0.1%甲酸可有效改善峰型,符合检测要求。
3结论
以色酮-3-甲醛为原料,首先以78%的收率制得色酮-3-甲腈,后经水解制备了色酮-3-甲酸,该步反应收率为90%,两步反应总收率为70%。
通过研究确定了水解反应的最佳条件:
乙二醇做溶剂、氢氧化钾与色酮-3-甲腈物质的量比为5∶1、氢氧化钾浓度为9.7mol·L-1、反应温度120℃、反应时间5h。
通过高效液相色谱检测产物纯度为98.17%,符合市场要求。
本方法避免了危险化学品的使用,具有收率高、易操作的优点,为色酮-3-甲酸的合成提供了新的思路。
参考文献:
[1]MPITIMPITIAN,PETZERJP,PETZER,etal.Synthesisandevaluationofchromonederivativesasinhibitorsofmonoamineoxidase[J].MolecularDiversity,2019,23(4):
897-913.
[2]SZULAWSKA-MROCZEKA,SZUMILAKM,SZCZESIOM,etal.Synthesisandbiologicalevaluationofnewbischromonederivativeswithantiproliferativeactivity[J].ArchivderPharmazie,2013,346
(1):
34-43.
[3]王晓丽,严丽,褚朝森,等.二氢色原酮-3-甲醛的合成工艺研究[J].当代化工,2019,48(3):
501-504.
[4]MILIUTINAM,EJAZSA,IAROSHENKOVO,etal.Synthesisof3,3'-carbonyl-bis(chromones)andtheiractivityasmammalianalkalinephosphataseinhibitors[J].Organic&BiomolecularChemistry,2015,14
(2):
495-502.
[5]TALHI,OUALID,PINTO,etal.Synthesisof5-(2-hydroxybenzoyl)-1,3-disubstituteduracils[J].Synlett.,2013,24:
1147-1149.
[6]曹玲华,张林,顾军.3-(3’-芳基-5’-硫酮-1’,2’,4’-三唑-4’-基)-氨甲酰基色酮类化合物的合成[J].有机化学,2002,22(6):
405-410.
[7]TARUNG,RANJANP,CHANDRAKANTAB.Studyofdifferencesinthereactivityofalkylandarylnitronesderivedfrom4-oxo-4H-1-benzopyran-3-carboxaldehyde[J].JournalOfchemicalresearch,2004
(1):
47-49.
[8]AMBARTSUMYANAA,VASILEVATT,CHAKHOVSKAYAOV,etal.Transformationsof4-Oxo-4H-chromene-3-carbaldehydeundertheactionofFe(CO)5[J].RussianJournalofOrganicChemistry,2012,48(3):
451-455.
[9]YOSHIMASAM,SEIICHIRON,SHIGETON,etal.Oxidationof4-Oxo-4H-1-benzopyran-3-carboxaldehydeswithN-bromosuccini-mide[J].SyntheticCommunications,1980,10(12):
889-895.
[10]REDDYGJ,LATHAD,THIRUPATHAIAHC,etal.Anefficientonestepconversionof3-Formylchromonesinto3-Cyanochromones[J].OrganicPreparationsandProceduresInternational,2004,36(3):
287-28.
[11]王晓丽,褚朝森,胡玉涛,等.1-苯基环丙基甲腈及衍生物的合成[J].精细化工中间体,2017,47(5):
28-31.
[12]HUANGA,MORETTOA,JANZK,etal.Discoveryof2-[1-(4-chlorophenyl)cyclopropyl]-3-hydroxy-8-(trifluoromethyl)
quinoline-4-carboxylicacid(PSI-421),ap-selectininhibitorwithimprovedpharmacokineticpropertiesandoralefficacyinmodelsofvascularinjury[J].JournalofMedicinalChemistry,2010,53(16):
6003-6017.
[13]SYLVESTERK,RICHARDEB,MAXIMILIANVS,etal.Substitutedchromone-3-carbonitriles,carboxamidesandcarboxylicacidusefulforpreventingasthmaticsymptoms:
UnitedStates,US3937837[P].1975-01-21.
[14]董乙文,严丽,王晓丽,等.3-(2-噻吩)丙烯酸的合成工艺研究[J].当代化工,2016,45(11):
2517-2519.
[15]褚朝森,王曉丽,彭飞宏,等.3-(2-噻吩)丙烯酸的合成工艺优化[J].四川化工,2018,21(6):
11-14.
[16]冯里,王晓丽,褚朝森,等.超声波相转移催化苯乙腈的环丙烷化反应[J].当代化工,2017,46(9):
1768-1770.
 升级会员
升级会员 