功能基因的序列比对方法.docx
《功能基因的序列比对方法.docx》由会员分享,可在线阅读,更多相关《功能基因的序列比对方法.docx(15页珍藏版)》请在冰豆网上搜索。
功能基因的序列比对方法
功能基因的序列比对
<1>.切除载体和(或)引物
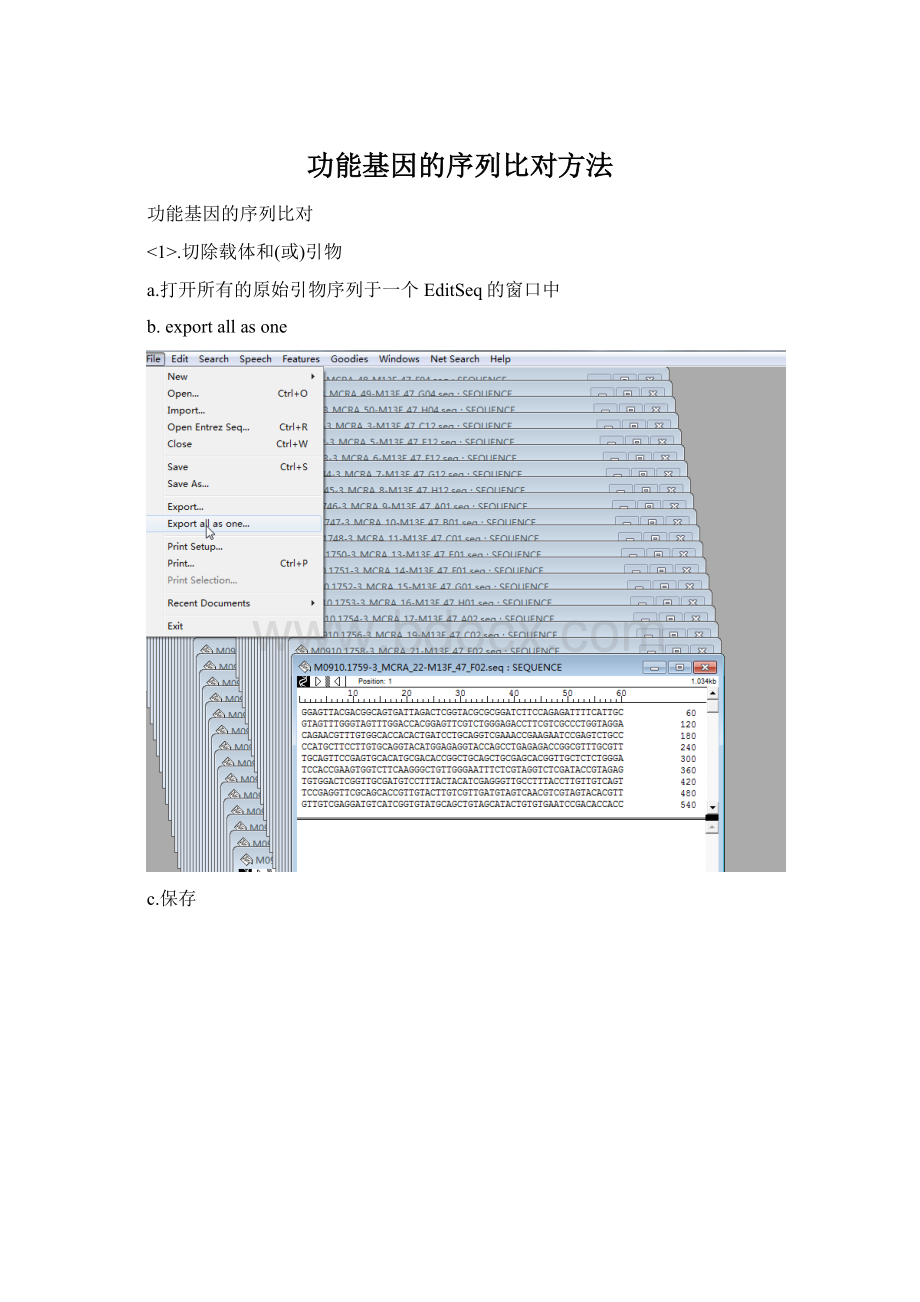
a.打开所有的原始引物序列于一个EditSeq的窗口中
b.exportallasone
c.保存
d.打开这个保存的文件,开始切除载体和引物
e.选择载体插入点两侧的序列(10-15个的样子)搜索注意:
不存在正反向的问题,都是一个方向,因为测序的时候是选择两个载体上的引物其中的一条来往后测序的!
切完之后另存为
f.重新打开这个文件,开始切除引物
方法同切载体,但是要注意正反向的问题。
比如mcrA基因,其引物为
Forward:
5'-GGTGGTGTMGGATTCACACARTAYGCWACAGC-3'
Reverse:
5'-TTCATTGCRTAGTTWGGRTAGTT-3'
先找Forward5’端,此时只找到的部分序列。
切去5’端。
然后再切这些切掉5’端序列的3’端的序列,此时其3’端序列应该是Reverse的反向互补序列。
切去这个反向互补序列,这样一来这个些序列就已经被切去两端的引物了。
但此时还剩下另一部分未切除任何引物的序列,此时记下这些序列的编号,先切去Reverse5’端。
再用Forward的反向互补序列切去3’端,这样剩下的序列也都被切除两端的引物了。
<2>将所有序列调整为同向序列:
a.选择前面记录编号的序列,将这些序列一个个都转换为其反向互补序列。
这样一来所有的序列都成为同向序列了,即在DNA两条反向互补链的其中一条上的比较了。
b.保存该文件
<3>生成OTUs
Google搜索”FastgroupII”
或fastgroup.sdsu.edu/fg_tools.htm
(Onlinegrouping--注意勾选的选项)
Choosemethod里面相似度可以选97%或98%
提交之后出现的窗口如
可以看到被分为了10个OUT每个OUT都自动选择了一个代表序列。
全选将其复制到word中,备用。
并把其中的那些代表序列都复制下来粘贴到TXT保存。
<4>寻找嵌合体:
一般是对16SrRNA来说的
两个:
decipher.cee.wisc.edu/FindChimerasOutputs.html(或搜decipherchimera)
comp-bio.anu.edu.au/bellerophon/bellerophon.pl(或搜bellerophonchimeracheck)
<5>翻译
:
pfam.sanger.ac.uk/
在保存有OTUs的TXT文件中,一个一个翻译成蛋白质序列。
最后保存。
在用Expasy翻译的时候选择第二个选项
点击翻译
理想的情况是这段序列中应该是没有终止序列的即”-”符号,因此先选择阅读框较长,整段序列也没有终止子的那些,如图,先选择第二个。
复制红色的区域,在blast上比对,看是否是需要的序列,如果是。
那么就选择此结果,如果不是,再一一比对其他的罗列结果。
或者直接将DNA序列提交到sanger上,出现如下结果
Frame2中有一段绿色,显示就是mcrA的保守家族。
那么Frame2即为正确的翻译方法。
另存为,只保留pro的序列的TXT
改名为.FAST格式
<6>寻找最相似序列
打开这个FAST文件,开始一个个找最相似序列了。
在这个窗口,开始blast。
找到一个序列后复制其DNA的编号
点击这个按钮
出现这个窗口
把复制的DNA编号手动输入点击OK则这个序列被自动添加到了FAST文件里了。
一般一个序列寻找3个相似度不等的序列。
最后,保存为一个新的FAST文件。
<7>画系统发育树
打开前面的FAST文件,全选文件”W”一下,再直接点OK
左右两头各删除带*之前的序列,另存为新的FAST文件。
打开这个FAST文件开始画树。
<8>最后对画的树进行一些修饰。
 升级会员
升级会员 