动植物基因组denovo常见问题.docx
《动植物基因组denovo常见问题.docx》由会员分享,可在线阅读,更多相关《动植物基因组denovo常见问题.docx(8页珍藏版)》请在冰豆网上搜索。
动植物基因组denovo常见问题
动植物基因组denovo常见问题
基础知识
1、什么是基因组denovo测序
答:
对某一物种进行高通量测序,利用高性能计算平台和生物信息学方法,在不依赖于参考基因组的情况下进行组装,从而绘制该物种的全基因组序列图谱。
2、普通基因组的定义
答:
单倍体,纯合二倍体或者杂合度<%,且重复序列含量<50%,GC含量为35%到65%之间的二倍体。
3、复杂基因组的定义
答:
杂合率>%,重复序列含量>50%,GC含量处于异常的范围(GC含量<35%或者GC含量>65%=的二倍体,多倍体。
诺禾致源对二倍体复杂基因组进一步细分为微杂合基因组(%<杂合率<%=、高杂合基因组(杂合率>%)以及高重复基因组(重复序列比例>50%)。
4、怎么查询基因组的大小
答:
查询植物基因组大小的网站:
;
查询动物基因组大小的网站:
。
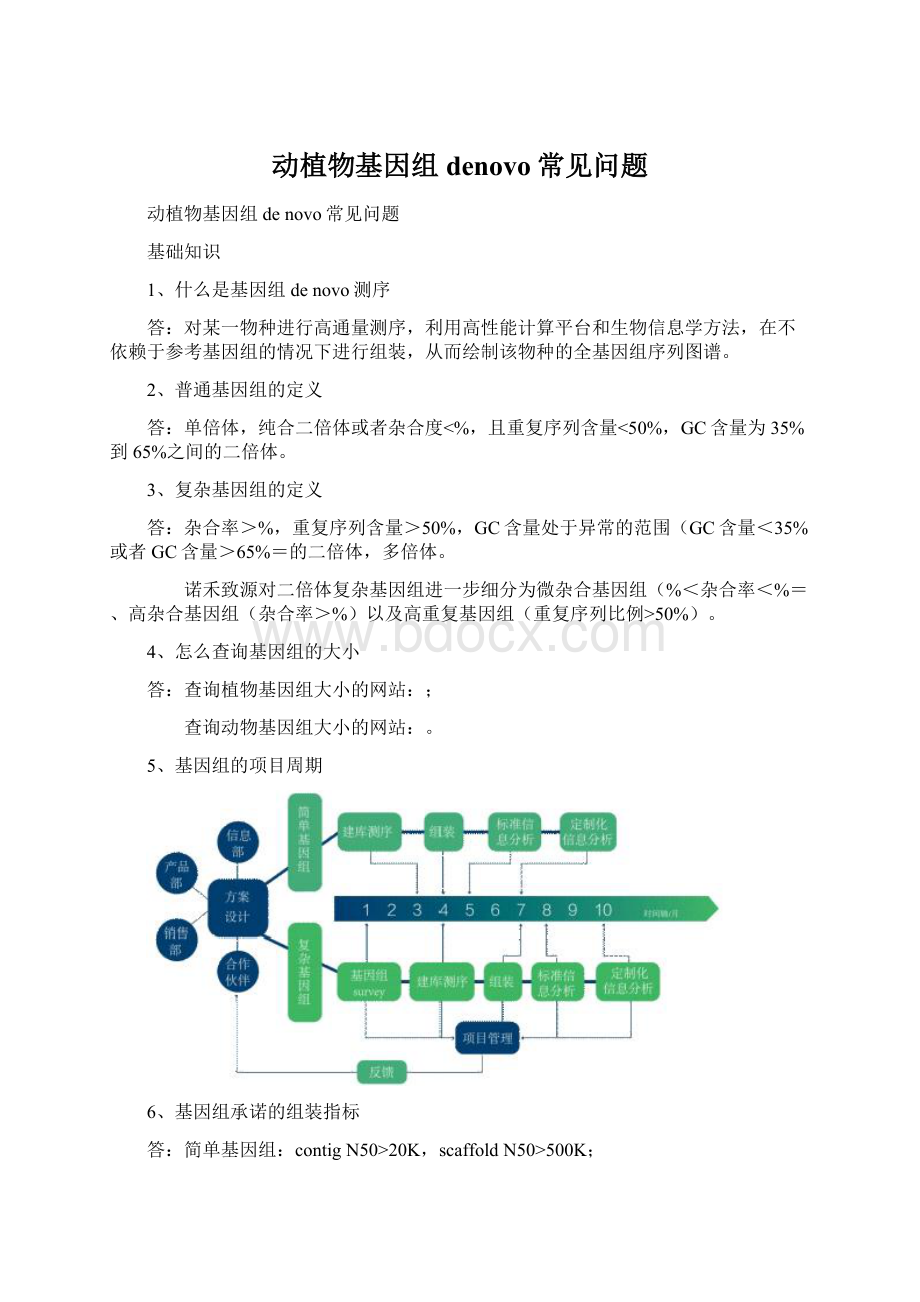
5、基因组的项目周期
6、基因组承诺的组装指标
答:
简单基因组:
contigN50>20K,scaffoldN50>500K;
复杂基因组:
contigN50>20K,scaffoldN50>300K。
样品要求
1、动植物基因组测序对取样有什么要求
答:
植物:
需要黑暗无菌条件下培养的黄化苗、组培苗,基因组样本量500μg~1mg,越多越好。
选择纯合或杂合度尽可能小的样品(杂合度<%)。
动物:
应选取肌肉、血液等含脂肪较少的部位取样,尽量选择同一个体取样,以减少个体差异性对后续拼接的影响。
基因组样本量500μg~1mg,越多越好。
样本的性别决定模式是XY型,则尽量选择雌性个体(XX型),如果是ZW型,则尽量选择雄性个体(ZZ型)。
2、全基因组测序对DNA样本有什么要求
答:
(1)样品需求量(单次):
小片段文库,≥3μg;2Kb~5Kb大片段文库,≥20μg;10Kb~20Kb大片段文库,≥60μg;完成全基因组测序样品DNA量需求约为500μg~1mg;
(2)样品浓度:
对于小片段文库,≥50ng/μl,对于2Kb~5Kb大片段文库,≥150ng/μl;对于10Kb~20Kb大片段文库,≥150ng/μl;
(3)样品纯度:
OD260/280=~;无蛋白质、RNA污染或肉眼可见杂质污染;
(4)样品质量:
基因组完整。
如需建立≥5Kb的插入片段文库,则电泳结果,基因组DNA主带≥23Kb;脉冲场电泳结果,基因组DNA主带≥40Kb。
文库构建
1、基因组测序的文库构建及测序策略
答:
简单基因组:
180bp、500bp、2K、5K、10K;PE100测序;测序深度一般为100-150X;
复杂基因组:
180bp、300bp、500bp、2K、5K、10K、20K;PE100测序;测序深度一般为200-300X。
2、DNAFragment文库的定义、用途及实验流程
答:
(1)定义:
将基因组或大片段DNA随机打断成<800bp的小片段(主要为200bp、300bp、500bp等),加上特定接头做成DNA文库后直接对DNA片段进行单末端(Single-End)或者双末端(Paired-End)测序,不需要克隆到细菌中,可以获得大量的DNA序列信息。
(2)用途:
DNAFragment文库制备的整个过程只需2天,单末端测序长度可达100bp,双末端为200bp。
该技术测序通量高,可在全基因组水平上最大限度的、完整的获取基因组及多态性信息。
广泛地应用于基因组的denovo测序、基因组重测序、BAC测序和长片段PCR产物测序等。
(3)实验流程:
3、DNAmate-pair文库的定义、用途及实验流程
答:
(1)定义:
首先将基因组DNA随机打断到特定大小(2-20kb);然后经末端修复,生物素标记和环化等实验步骤后,再把环化后的DNA分子打断成400-600bp的片段并通过带有链亲和霉素的磁珠将带有生物素标记的片段捕获。
这些捕获的片段再经末端修饰和加上特定接头后建成大片段文库,不需要克隆到细菌中,直接在Illumina测序仪上进行测序。
通过大片段文库构建,从而获得基因组中较大跨度(2-20kb)片段两端的序列。
(2)用途:
DNAMate-pair文库制备的整个过程需要5天,这种从较大跨度两端所获得的序列对基因组denovo项目的组装和基因组结构变异发掘具有非常重要的作用。
(3)实验流程:
信息分析
1、什么是Read、Contig、Scaffold
答:
Read:
测序读到的碱基序列片段,测序的最小单位;
Contig:
由reads通过对overlap区域拼接组装成的没有gap的序列段;
Scaffold:
通过pairends信息确定出的contig排列,中间有gap。
2、什么是N50,N70,N90
答:
把组装出的contigs或scaffolds从大到小排列,当其累计长度刚刚超过全部组装序列总长度50%时,最后一个contig或scaffold的大小即为N50的大小,N50对评价基因测序的完整性有重要意义;N70和N90的计算方法与N50类似,只是百分数变为70%或90%。
3、普通基因组的解决方案
答:
诺禾采用自主升级的SOAPdenovoII进行普通基因组组装。
组装流程(图1)包括:
(1)构建不同长度的插入片段文库;
(2)构建deBrujin图;
(3)化简deBrujin图;
(4)构建contigs;
(5)构建scaffolds;
(6)补gaps;
诺禾致源的技术升级包括:
(1)开发了新的序列纠错模块,降低测序错误对组装的影响;
(2)在contigs组装步骤,开发了StepK连接模块,以混合拼接算法连接contigs,从而提升原始的contigs长度;
(3)在scaffolds组装步骤,开发了ctgdistanceevaluation模块,更精确地评估contigs间的距离;同时开发了scafconstruction模块,以新的连接单位来组装scaffold,从而提升scaffolds的连接准确率及长度。
图1 基因组denovo测序及拼接组装流程
经过以上几步,最终简单基因组的组装结果至少应达到contigN50>20K,scaffoldN50>300K。
4、复杂基因组(二倍体杂合)的解决方案
答:
针对复杂基因组中二倍体杂合基因组,诺禾致源开发了NOVOheter软件,成功实现了二倍体杂合基因组组装。
与SOAPdenovo相比,NOVOheter软件组装二倍体杂合基因组的技术创新主要体现在以下几个方面:
(1)通过高深度测序(200-300X)将基因组上的杂合和纯合区域分开;
(2)利用reads信息和PE关系连接杂合位点,延长原始contigs:
在杂合部分间距离较短的情况下,利用reads信息将杂合位点连接起来,若杂合部分间距离较长时,利用Pair-End关系连接杂合位点(所以需要加入更多类型的小片段文库,以连接不同距离的杂合位点),从而提高了contigs的长度,为后续组装打下基础(图3);
图3基于NOVOheter软件构建contigs
a:
利用深度信息区分杂合部分(覆盖度为n)和纯合部分(覆盖度为2n);
b:
若杂合部分的距离较短(如60bp),则可利用reads信息将杂合位点连接起来;
c:
若杂合部分的距离较长(如400bp),则利用Pair-End关系,将杂合位点连接起来;
d:
得到杂合contigs。
注:
图中不同颜色的点表示杂合位点。
(3)分区域构建scaffolds:
同样利用contigs深度信息区分纯合contigs和杂合contigs;利用Pair-End关系将纯合contigs,杂合contigs分别组装成scaffolds;最后将相邻的纯合contigs和杂合contigs进行连接,构建更长的scaffolds。
5、如何评价组装结果
答:
常染色体区的覆盖度:
评价基因组常染色体区的覆盖度,可以用BAC或者是Fosmid序列来评估;把已公布或者客户提供的BAC或fosmid克隆序列作为Refrence,将拼接完成的基因组序列map回已知的BAC或者fosmid序列上,检查拼接的序列对已知序列的覆盖度到什么水平。
基因区的覆盖度:
评价基因区的覆盖度,可以用EST序列或者是转录组序列来评估;把已公布或者客户提供的EST或转录组序列作为query序列map到拼接完成的基因组序列上,检查拼接序列对已知序列的覆盖度是达到什么水平。
6、影响基因组组装的因素
答:
基因组的重复序列和杂合度,是否污染以及基因组的倍性情况。
7、基因组项目的标准生物信息分析的内容
答:
基因组项目的标准生物信息分析的内容如下:
(1)数据处理;
(2)基因组组装:
基因组评估:
基因组大小、GC含量、复杂序列、杂合度评;
组装:
数据纠错;Contig、Scaffold组装;Gap填充;组装质量分析、评估和结果统计;
(3)基因组注释:
重复序列注释;基因预测;基因组功能注释;非编码RNA注释;
(4)比较基因组学分析:
基因家族鉴定;
基因组共线性分析;
全基因组复制分析(动物:
WGAC;植物:
WGD);
正选择基因的鉴定及功能分析;
基因家族的扩增收缩分析;
系统发育分析;
物种分化时间估。
 升级会员
升级会员 