酮洛芬的工艺合成.docx
《酮洛芬的工艺合成.docx》由会员分享,可在线阅读,更多相关《酮洛芬的工艺合成.docx(7页珍藏版)》请在冰豆网上搜索。
酮洛芬的工艺合成
酮洛芬的工艺合成
1产品简介
1.1中英文名称,分子式,结构式
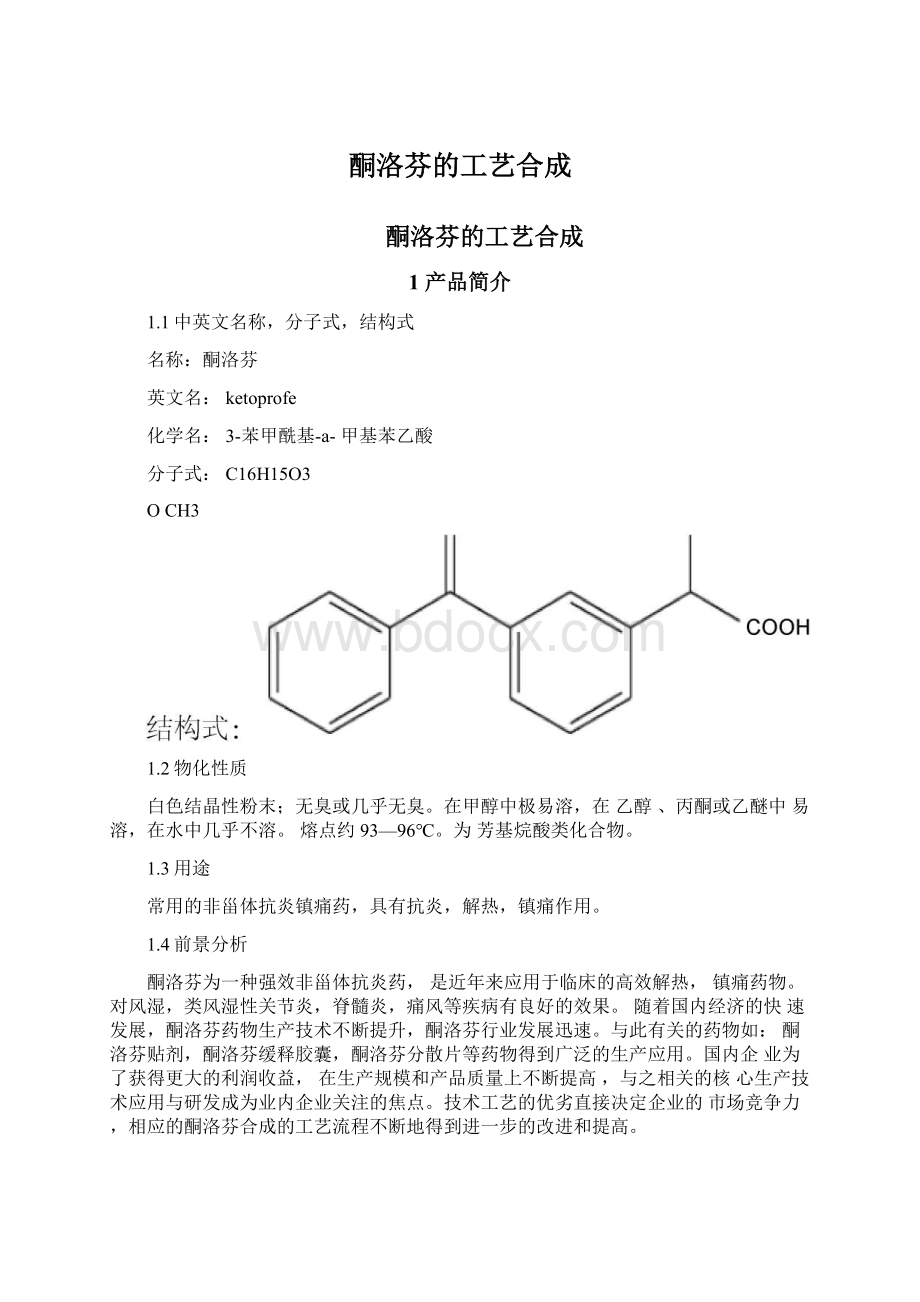
名称:
酮洛芬
英文名:
ketoprofe
化学名:
3-苯甲酰基-a-甲基苯乙酸
分子式:
C16H15O3
OCH3
1.2物化性质
白色结晶性粉末;无臭或几乎无臭。
在甲醇中极易溶,在乙醇、丙酮或乙醚中易溶,在水中几乎不溶。
熔点约93—96℃。
为芳基烷酸类化合物。
1.3用途
常用的非甾体抗炎镇痛药,具有抗炎,解热,镇痛作用。
1.4前景分析
酮洛芬为一种强效非甾体抗炎药,是近年来应用于临床的高效解热,镇痛药物。
对风湿,类风湿性关节炎,脊髓炎,痛风等疾病有良好的效果。
随着国内经济的快速发展,酮洛芬药物生产技术不断提升,酮洛芬行业发展迅速。
与此有关的药物如:
酮洛芬贴剂,酮洛芬缓释胶囊,酮洛芬分散片等药物得到广泛的生产应用。
国内企业为了获得更大的利润收益,在生产规模和产品质量上不断提高,与之相关的核心生产技术应用与研发成为业内企业关注的焦点。
技术工艺的优劣直接决定企业的市场竞争力,相应的酮洛芬合成的工艺流程不断地得到进一步的改进和提高。
2合成方法
2.1第一种合成方法
(1)合成原理
以苯甲酸为起始原料,经溴化、Friedel-crafts反应、Grignard反应制得3乙酰基二苯酮,再经Darzens反应制得酮基布洛芬。
(2)合成所需要的原料
苯甲酸,铁粉,溴,五氯化磷,无水三氯化铝,镁粉,醇钠的乙醇溶液,氯乙酸乙酯乙腈,无水四氢呋喃
(3)生产工艺,包括原料的用料比例,反应条件等。
第一步:
3-溴苯甲酸的制备
在干燥的四颈瓶中,加入73.2g(0.6mol)化合物苯甲酸、4g还原铁粉,于100℃~150℃搅拌0.5h,滴加18mL(0.35mol)溴,加毕搅拌1h,追加4g铁粉和18mL溴,于150℃搅拌2h后,再于260℃搅拌3h,将产物溶于稀碱中,过滤后,用盐酸酸化使其析出晶体,重结晶,干燥得白色粉末固体
第二步:
3-溴二苯甲酮的制备
在干燥的三颈瓶中,依次加入化合物3-溴苯甲酸55g(0.275mol)、五氯化磷59.8(0.333mol)及无水苯187.5mL,于80℃搅拌回流2h,用冰盐浴冷却至5℃以下,缓慢加入无水三氯化铝71.9g(0.95mol),温度控制在10℃以下,约1h加完,在45~60℃搅拌回流5h,冷至室温后,将反应液倾入冰(500g)和浓盐酸(200mL)的混合液中,分出有机层,水层用苯提取(100mL×2).合并有机层,依次用饱和碳酸氢钠溶液200mL和水(200mL×2)洗至中性,无水硫酸镁干燥,常压回收溶剂,得淡黄色固体70g。
第三步:
3-乙酰基二苯酮的制备
滴加在干燥的四颈瓶中,依次加入镁粉1g,无水四氢呋喃25mL和少许碘粒.加热搅拌至60~100℃时,11g(0.04mol)化合物3-溴二苯甲酮和无水四氢呋喃35mL的混合液,加毕,搅拌2.5h,冷至室温,滴加乙腈55mL(0.086mol)和无水四氢呋喃15mL的混合液,加毕搅拌4h,回流3h,减压回收溶剂,加20%盐酸100mL,回流3h,放置过夜,加入三氯甲烷100mL,搅拌,静置,分出有机层,依次用饱和碳酸氢钠溶液(100mL×2)和饱和氯化钠溶液(100mL×2)洗至中性,无水硫酸镁干燥,回收溶剂后,得黄色液体10g。
第四步:
酮洛芬的制备
在三颈瓶中将11.2g(0.05mol)化合物3-乙酰基二苯酮和8g(0.05mol)氯乙酸乙酯溶于50mL乙醇中,边搅拌边滴加醇钠的乙醇溶液30mL,回流2h,加入30%氢氧化钠溶液40mL,搅拌2h,用盐酸调至pH5,回流1h,加重铬酸钾和硫酸的水溶液(1∶4),滴至变绿为止.用苯-石油醚(V∶V=3∶10)萃取上述液,分出有机层,
减压回收溶剂,得黄棕色油状物,放置固化,用苯-石油醚重结晶,得白色晶体7g。
(4)
工艺流程图
(2)合成所需要的原料:
腈乙基二苯甲酮、乙二醇、氢氧化钾、盐酸、丙酮
(3)生产工艺,包括原料的用料比例,反应条件等。
第一步:
酮洛芬粗品的合成
在三口烧瓶中投入腈乙基二苯甲酮5g和乙二醇5mL,加热搅拌使之完全溶解。
在烧杯中加入3g氢氧化钾,加入5mL纯化水,搅拌至溶解完全。
待腈乙基二苯甲酮溶液升温至150℃,将配好的氢氧化钾溶液倒入反应器中控制温度150℃反应45min,原料全部转化为K盐,待反应液冷却,加入60mL纯化水稀释,继续搅拌,待用。
用试剂盐酸将制得的K盐水溶液;pH调至8~9,加入0.5g活性炭,搅拌加热30min,趁热过滤;配置0.5mol/L的盐酸水溶液150mL,将滤液缓慢滴加到稀盐酸溶液中,并搅拌,先慢后快直至滴加结束。
将析出的固体过滤,滤饼放入烘箱干燥,得粗产物5.3g。
第二步:
产品纯化
将所得的粗产品溶于10mL丙酮,搅拌直至全部溶解,加入0.5g活性炭,加热脱色30min,趁热过滤。
(4)工艺流程图
(5)
氢氧化钾溶液
酸
(6)产率计算,成本核算:
收率达99.1%。
2.3第三种合成方法
(1)合成原理
以3-氰甲基苯甲酸为起始原料,经过Friedel-Crafts、单甲基化、水解3
步得酮洛芬。
(2)合成所需要的原料
对硝基苯乙酮,碳酸二甲酯,三乙基氯化铵,亚硫酰氯,苯,氯化铝,碳酸钾等。
(3)生产工艺,包括原料的用料比例,反应条件等
第一步:
5-(2-甲基-1,3-二噁烷-2-基)-7-苯基苯并[c]异噁唑(3)的制备
对硝基苯乙酮(100g,0.6mol)苯(500ml)、乙二醇(120ml,2.2mol)、对甲苯磺酸(1.5g,7.9mmol)回流带水15h,加入苯(100ml),静置30min,下层(醇层)趁热用苯(20ml)提取,合并苯层,减压回收苯后降至35℃,加入含氢氧化钠(124g)的95%乙醇(500ml),搅拌,30min内于45℃滴加苯乙腈(74ml,0.6mol),滴毕,于57~60℃反应7h,室温静置过夜。
搅拌下加水(350ml)后继续搅拌30min,过滤,滤饼用水洗至中性,80℃干燥后得到浅黄色固体(3)。
第二步:
2-氨基-5-乙酰基二苯甲酮(4)的制备
3(50g,0.2mol)、95%乙醇(400ml)和36%盐酸(8ml,0.1mol)升温至60~70℃,保温搅拌30min后降至40℃以下,加60~80目铁粉(47g,0.9mol),缓慢升温回流反应2h,降至70℃,用固体氢氧化钠(约3.7g)调至pH7.2~7.5,趁热过滤,滤液减压回收乙醇后降至室温析晶,过滤,滤饼用少量乙醇和水洗后于80℃烘干,得黄色固体4(39.1)g。
第三步:
3-乙酰基二苯甲酮(5)的制备
4(26.0g,0.1mol)溶于乙醇(60ml)和浓硫酸(14ml),15℃搅拌下慢慢加入亚硝酸钠(8.5g,0.1mol)的水溶液(100ml),装上冷凝器后缓慢升温,有大量氨气逸出,改水浴为油浴、回流装置为分馏装置,小心蒸除乙醇及乙醛,汽温达80℃时停
止。
降温后用苯(50ml)提取,用少量水和5%氢氧化钠溶液洗后再用水洗至中性。
无水氯化钙干燥后常压蒸除溶剂,减压收集152~156℃/30Pa馏份,得浅黄色液体5g。
第四步:
酮洛芬的合成(6)的制备
在装有冷凝管、机械搅拌器的500mL三颈烧瓶中,加入3-苯甲酰基-a-甲基苯乙腈50g(0.212mol)、冰醋酸50mL,49%硫酸溶液100mL(浓硫酸:
水=1:
1),搅拌升温并保持在125-130度之间回流4.5h后,逐渐降至40度加入水150mL,冷却至室温后加
甲苯50mL,搅拌后,静置分层,分出有机相,水相用甲苯萃取50mL×2,合并有机相。
有机相水洗至中性,用10%KOH溶液调至pH值>10以上,静置分层,回收甲苯,水相用活性炭脱色后过滤,滤液先用36%乙酸调至中性,再用10%盐酸调至pH值<2,析出白色固体,干燥得粗品50.5g定纯度为98.5%,用苯,石油醚重结晶,过滤,干燥得白色固体(6),称重48.9g。
(4)工艺流程图
3优缺点评价
第一种合成方法:
反应过程简单,易操作,产率高,但反应时间较长。
第二种合成方法:
本工艺以腈乙基二苯甲酮为原料,经碱性条件下水解得酮洛芬反应时间从10h缩短为1h,副反应少,原料转化率高,产品收率达温度过低反应速率偏低,反应时间过长,能耗较大,温度过高,有明显的自身缩合副产物生成(高于150℃就有明显的副产物生成)溶剂对该反应有着重要的影响乙二醇溶解性及反应性较好,且乙二醇与水互溶,后处理非常简便,本方法可以供工业化生产。
第三种合成方法:
新甲基化试剂碳酸二甲酯为代替传统的CH3I,CH3Br,(CH3)2SO4等甲基化试剂,副产物少,有利于产物的分离纯化;同时避免了CH3I,CH3Br,(CH3)2SO4的剧毒性,反应条件温和,改善了工作环境;以3-氰甲基苯甲酸为起始原料,经Friedel-Crafts反应、a-单甲基化,水解3步反应制得酮洛芬,合成路线短、收率高、操作简单等优点,适合大生产。
总体来说第三种合成方法较好。
4酮洛芬的红外表征图谱
在酮洛芬红外光谱中在1600-1700/cm有二重峰为酮洛芬的N-H伸缩振动区
5参考文献
[1]陈芬儿,张文文.酮洛芬的合成[J].中国医药工业杂志,1991,22(8):
334-335.
[2]陈芬儿,张文文,董恩莲.酮洛芬的合成改进方法[J].中国医药工业杂志,1992,23(7):
290-292.
[3]余红霞,郭峰,陈芬儿..酮洛芬的合成改进方法[J].中国医药工业杂志,1992,13
(2):
97-98.
[4]吕布,汪永忠,李颖.酮洛芬的合成改进方法[J].中国药物化学杂志,2000,10
(2):
127-128.
[5]胡钊侠.酮洛芬的合成新工艺:
CN,1143624[P].1997-02-26.
[6]万江陵,冯秀梅,陈芬儿.羟基缩合法合成酮洛芬[J].华西药学杂志,1996,11(4):
221-222.
 升级会员
升级会员 