液相色谱进阶讲座之梯度洗脱Word下载.docx
《液相色谱进阶讲座之梯度洗脱Word下载.docx》由会员分享,可在线阅读,更多相关《液相色谱进阶讲座之梯度洗脱Word下载.docx(12页珍藏版)》请在冰豆网上搜索。
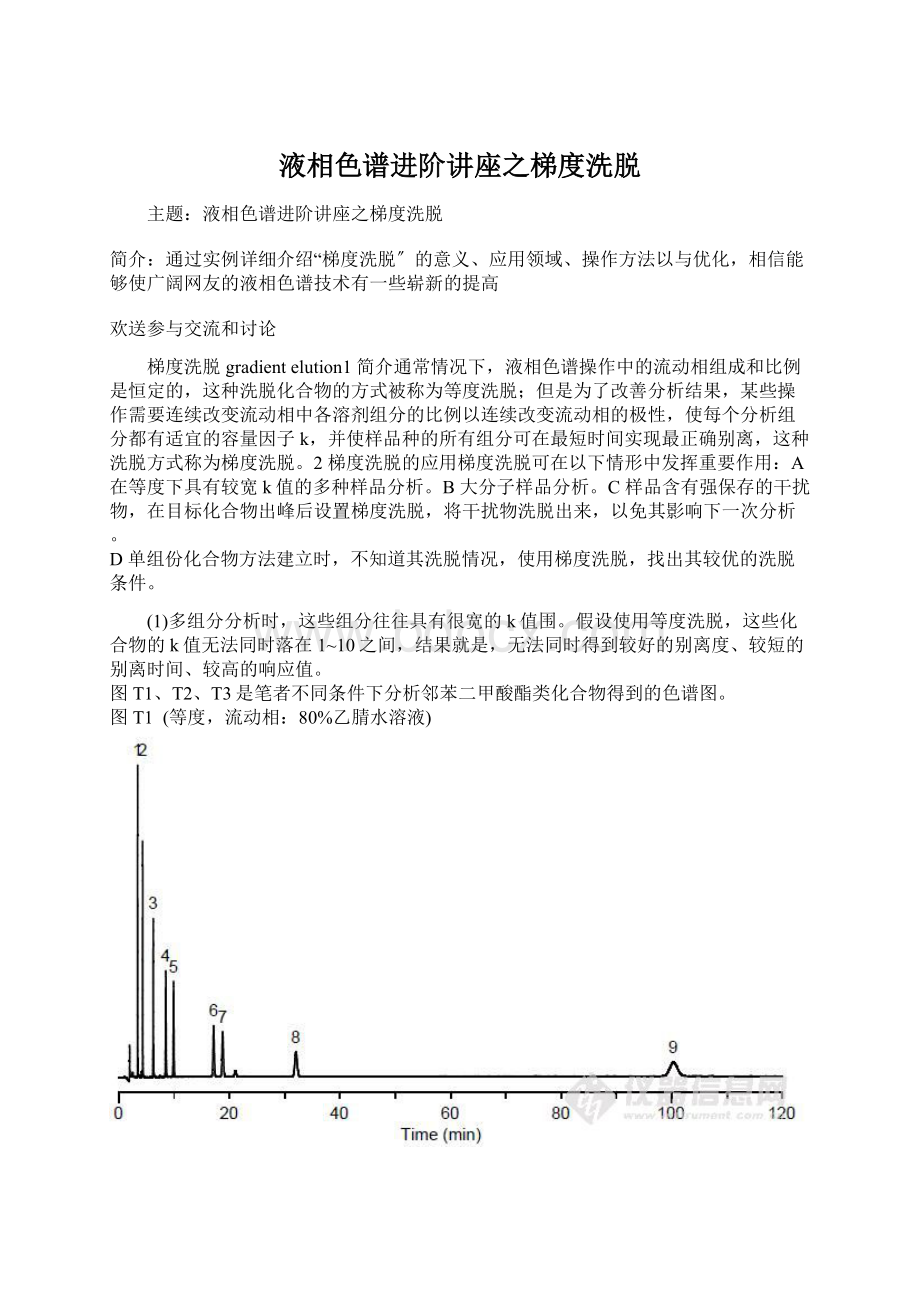
80%乙腈水溶液)
图T2(等度,流动相:
100%乙腈)
图T3(梯度,0-5min,乙腈由70%上升到90%,5-10min乙腈由90%上升到100%,10-18min,乙腈100%)
其他色谱条件:
色谱柱:
DiamonsilC18
(2),250*4.6mm,5μm流速:
1.0ml/min
检测器:
UV254nm
1邻苯二甲酸二甲酯(DMP)
2邻苯二甲酸二乙酯(DEP)
3邻苯二甲酸二正丙酯(DPrP)
4邻苯二甲酸丁基苄基酯(BBP)
5邻苯二甲酸二正丁酯(DBP)
6邻苯二甲酸二戊酯(DPP)
7邻苯二甲酸二环己酯(DCHP)
8邻苯二甲酸二己酯(DHP)
9邻苯二甲酸(2-乙基己基)酯(DEHP)
三种洗脱方式比照
条件
最小别离度
分析时间
灵敏度
等度1(T1)
>4
>100min
1-9,灵敏度逐渐降低,8和9难以检出
等度2(T2)
=0.8
11min
1-9,灵敏度逐渐降低,均较高
梯度(T3)
>3
25min
1-9,灵敏度相差不大,均较高
由该分析案例可知,在多组分分析时,只要能够选出适宜的梯度条件,就能够实现“较好的别离度、较短的别离时间、较高的响应值〞。
(2)样品溶液中同时含有目标化合物和强保存化合物,目标化合物流出后使用梯度洗脱将强保存化合物洗下来,以防止强保存化合物污染色谱柱或在后续的分析中流出干扰分析。
T4某分析项目末端梯度洗脱以洗掉过剩的衍生试剂(末端梯度:
39-40min,有机相含量由38%升高到80%,保持10min)
由T4可以看出,40min左右,最晚出峰的组分已经被洗下来,然而却把有机相在1min由38%升高到80%,目的就是为了洗脱衍生反响剩余的衍生试剂(50min处的色谱峰)。
前面的杂质峰为样品基质中的强保存干扰物,末端的梯度洗脱同样也将其洗了下来,这就防止了这些化合物在随后的分析中缓慢流出造成的基线起伏。
(3)当遇到新的化合物需要分析,我们没有文献可以参考,只能初步圈定分析模式(反相、正相等等),此时确定不了等度洗脱的流动相组成和比例,只好借助梯度洗脱。
3梯度洗脱的原理(以反相色谱为例)梯度洗脱的实质就是,样品随梯度起点的流动相进入色谱柱入口,由于起点流动相中强洗脱溶剂B含量较低,化合物C1(保存性适中)和化合物C2(保存较强)因k值较高而滞留于色谱柱入口附近(几乎未移动)。
一段时间后,B增加到一定数值,C1的k值到达足够小(比方小于10),其在柱中的移动速率明显增大,并且随着B的增加,其移动速率愈加快速,直到tC1时流出色谱柱,被检测器检测到并被记录成色谱峰C1,tC1虽然比等度时的保存时间长很多,但C1的的平均k值却很小,因而色谱峰并未展宽,灵敏度仍处于较高水平。
B持续增加,在随后的某个时间点,C2的k值也降到10以下,C2的移动速率也开场变快,以与C1类似的方式于tC2被洗脱出色谱柱,其平均k值同样很小,色谱峰亦未展宽,灵敏度处于较高水平。
T5梯度洗脱过程中的峰迁移
黑色虚线代表B比例的变化(对应左侧上方纵轴)
红色曲线代表化合物k值随流动相改变而发生的变化(对应右侧纵轴)
绿色曲线代表化合物化合物在柱中的位置变化(对应左侧下方纵轴)
紫色曲线为色谱图
4梯度别离的建立梯度洗脱可以按以下步骤建立;
A选择初始条件:
色谱柱(类型和规格)、流动相组成、流速、柱温等等B根据A的分析结果升高起始流动相中的B%并降低完毕流动相中的B%,以去除色谱图中第一个峰出现前的和最后一个峰出现后的无意义时间C当峰别离较差或运行时间过长的时候,应通过调整强洗脱溶剂B、B升高速率或使用分段梯度等方式予以解决D考察不同仪器对梯度别离的影响。
(1)选择梯度条件
流动相:
选择一种强洗脱溶剂和一种弱洗脱溶剂,反相中常用强洗脱溶剂为乙腈和甲醇,弱洗脱溶剂为水,(由于反相中别离的化合物许多具有解离性,所以水相往往有酸、缓冲盐等,这些不在本讲座讨论围以,本讲座提到的水相可能包括纯水、缓冲盐水溶液等,具体事例会明确写出)。
正相中常用的强洗脱溶剂为甲基叔丁基醚、异丙醇等,弱洗脱溶剂为正己烷。
流速:
对于4.6mm径的色谱柱一般把流速设定为1.0~1.5ml/min,其他径的色谱柱可以以此标准进展换算;
柱温:
如有柱温箱,设定柱温35~45º
C为宜;
初始变化速率:
可以按下面的公式计算确定
F为流速,单位为ml/min;
Vm为色谱柱死体积,单位为ml;
k*为期望的平均保存因子;
△%B为强洗脱溶剂的变动值;
tG为梯度时间,单位为min。
该公式适用于相对分子量处于50~500之间的化合物。
对于150*4.6mm的色谱柱,Vm约为1.2ml;
流速通常设定为1.0ml/min;
k*为5是比拟好的选择;
因此△%B/tG应为3.3,也就是B的改变速率设定为3.3%是适宜的,当梯度围为5%-100%,梯度时间可以设为29min。
梯度围:
为防止峰遗漏,初始梯度围最好设置为强洗脱溶剂变动围为5%-100%,在反相中可以把乙腈的变动围设置在5%-100%,如果是能够耐受纯水的色谱柱,也可以设定在0%-100%。
梯度形状:
在初始梯度实验中,梯度可设置成线性(不分段),在后续的调整中那么可以设置分段梯度,每一段梯度的B变动斜率存在差异。
(2)调整强洗脱溶剂B、梯度变化速率或使用分段梯度等方式以优化梯度条件流动相对选择性起着至关重要的作用,流动相组成或比例的改变均会显著改变别离度,在梯度中亦然。
反相模式梯度的初试中,强洗脱溶剂往往选择乙腈,但乙腈只是在某些项目中具有较好的选择性,而另一些项目甲醇或许更适宜。
乙腈、甲醇洗脱能力相近,但二者的选择性那么具有互补性。
T6、T7、T8是PITC柱前衍生-反相液相色谱分析18种氨基酸的色谱图。
T6以乙腈作为强洗脱溶剂,丙氨酸和脯氨酸(7和8)别离度缺乏1.5;
T7以甲醇作为强洗脱溶剂,9后面的组分别离很差。
上述说明,甲醇对前面一组峰的别离较好,儿乙腈那么更适合后面一组峰,因此笔者把甲醇乙腈混合液作为强洗脱溶剂,T8证明这种混合溶剂确实能够将18种天然氨基酸、标物正亮氨酸以与NH3的的别离度到达2.0以上。
T6B为80%乙腈水溶液
T7B为80%甲醇水溶液
T8B为甲醇:
乙腈:
水=20:
60:
20
其他色谱条件
DiamonsilAAA氨基酸分析柱,250*4.6mm,5μm;
流动相B:
见谱图上方的描述流动相A:
0.05mol/L乙酸钠水溶液(pH=6.5)流速:
1.0ml/min检测器:
UV254nm
35摄氏度
具体化合物:
1Asp(天冬氨酸);
2Glu(谷氨酸);
3Ser(丝氨酸);
4Gly(甘氨酸);
5His(组氨酸);
6Arg(精氨酸);
7Thr(氨酸);
8Ala(丙氨酸);
9Pro(脯氨酸);
10NH3;
11Tyr(酪氨酸);
12Val(缬氨酸);
13Met(蛋氨酸);
14Cys(胱氨酸);
15Ile(异亮氨酸);
16Leu(亮氨酸);
17Nle(正亮氨酸);
18Phe(苯丙氨酸);
19Trp(色氨酸);
20Lys(赖氨酸)
化合物列表与T8上的峰号严格对应。
梯度变化速率梯度变化速率与平均保存因子k*是成反比关系的,梯度变化速率降低,k*值增加,并会产生如下结果:
其一,各组分间的别离度R提高;
其二,色谱峰的区域宽度增加,灵敏度下降;
其三,分析时间延长。
第一点是积极的,第二、第三点是消极的。
所以应在别离度能满足要求的前提下,控制梯度变化速率不要过低。
下面是梯度变化速率的影响:
测试项目为15种农药,唯一变量为梯度变化速率。
T9
甲醇变化速率20%/min,k*=1
T10
甲醇变化速率5%/min,k*=4
T11
甲醇变化速率1%/min,k*=15
C18,250*4.6mm,5μm
甲醇,水
1.7ml/min
室温
由T9、T10、T11可知,随着强洗脱溶剂变化速率降低,各组分的别离度逐渐增大,分析时间也逐渐延长。
如果改变溶剂、调整梯度变化速率仍不能较好别离那么需要改成非线性梯度。
下面是偶氮释放的24种芳香胺分析:
T12
偶氮释放的24种芳香胺分析的梯度设置
B为甲醇,A为0.005mol/L磷酸二氢铵+0.005mol/L磷酸氢二钠水溶液
T14
色谱图
结合梯度和色谱图可知,1-6是由第一段梯度(蓝色虚线)洗脱下来,B升高速率为0.5%;
7-16由第二段梯度(粉色虚线)洗脱下来,B升高速率为0.27%,色谱峰宽度较大;
17-24由第三段梯度(绿色虚线)洗脱下来,B升高速率为1.85%,因而峰形最为锋利。
(3)不同仪器对梯度分析结果的影响以与消除影响的方法
与等度分析不同,梯度分析时,色谱柱入口的流动相状态并不能与梯度曲线上的流动相状态在时间上准确对应。
比方,0min流动相比例已经开场变化了,这种变化首先由泵或比例阀做出动作,而此刻柱入口处的流动相组成仍是初始流动相,变化的流动相穿过泵或比例阀到柱头之间的空间后才能传到达柱入口,所以色谱柱入口的流动相状态始终滞后于时间程序上的流动相状态。
滞后时间tD=VD/F,VD是泵或比例阀到柱头之间的体积,F为体积流速。
不管是高压梯度还是低压梯度,滞后体积通常处于2~8mL之间,滞后体积的不同会导致以下现象:
其一,在不同款式仪器上使用一样梯度,保存时间会发生变化;
其二,在一种款式的仪器上开发的梯度洗脱方法换到另一款仪器上,可能会出现别离度下降、峰位置变化等现象
可用用下面的方法消除上述不利影响:
在A仪器上(VD=V1)开发梯度方法,在开发的梯度程序前预先参加5min等度洗脱(比例与初始流动相比例一样),并确定这5min的等度为影响别离,这就可以作为最终的梯度条件;
等换到B仪器上(VD=V2),假设V2=V1,梯度程序不需改变;
假设V2大于V1,那么应把前面的等度时间调小,减小值应为(V2-V1)/F;
假设V2小于V1,那么应把前面的等度时间调大,增加值应为(V1-V2)/F。
说到这里了,您可能会问我该怎样获得仪器准确的滞后时间呢?
您可以这样测定:
将色谱柱卸下,用无体积两通将色谱柱入口管线与出口管线连接。
以甲醇做A相,0.1%丙酮做B相,流速为1ml/min,先以B=0%的流动相平衡一段时间,响应值调为0,然后运行梯度,B20min由0%增到100%,并且在B为100%的条件下平衡20min,数据采集到40min左右。
从色谱图上找出,响应值到达(最大值-最小值)/2时的保存时间tx,tD=tx-10,VD=TD*F。
具体如以下图。
讲座局部到此处完毕,欢送大家踊跃参加讨论。
 升级会员
升级会员 