固相萃取基本原理与操作Word格式文档下载.docx
《固相萃取基本原理与操作Word格式文档下载.docx》由会员分享,可在线阅读,更多相关《固相萃取基本原理与操作Word格式文档下载.docx(27页珍藏版)》请在冰豆网上搜索。
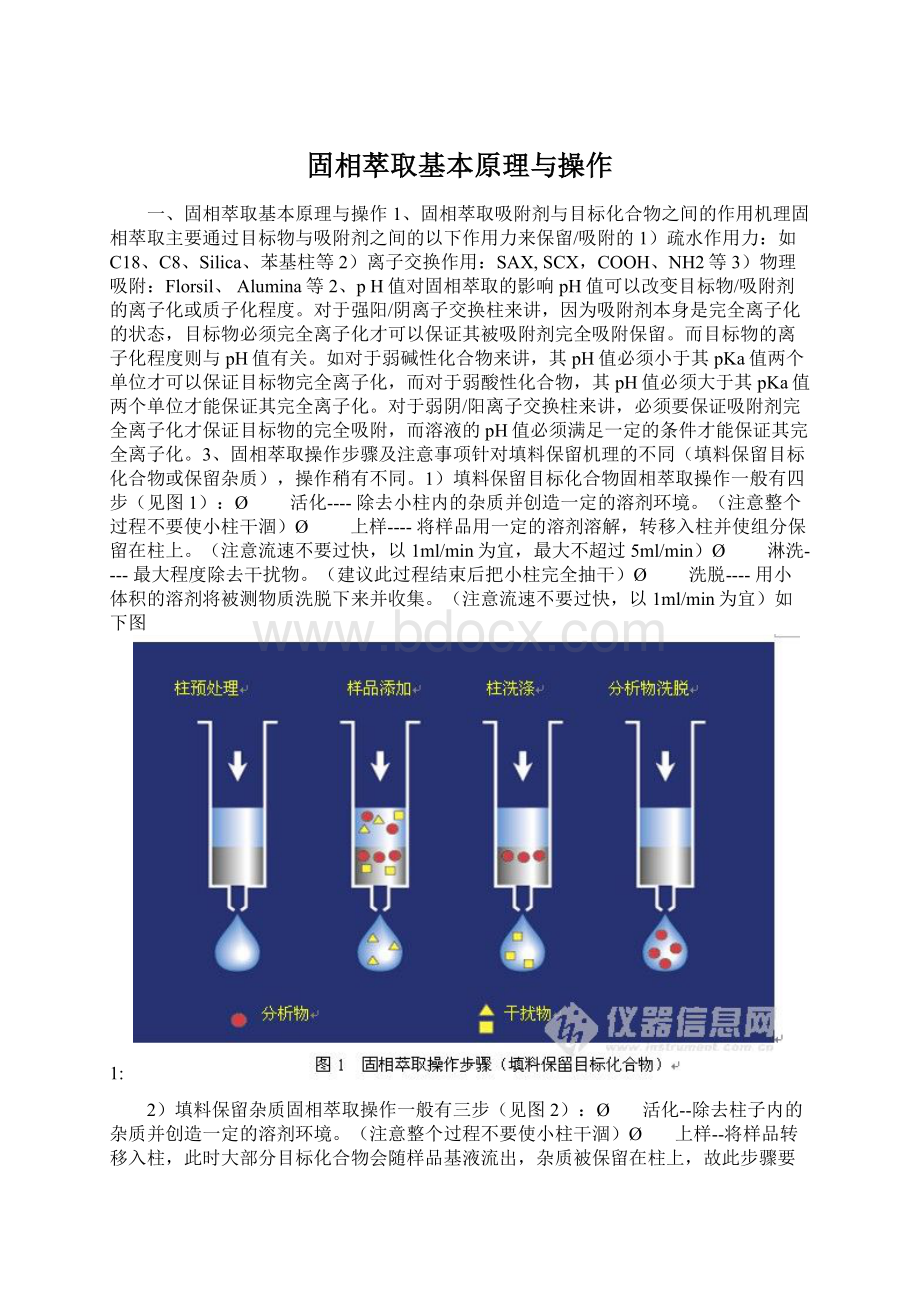
洗脱---用小体积的溶剂将组分淋洗下来并收集,合并收集液。
(注意流速不要过快)此种情况多用于食品或农残分析中去除色素。
如下图2:
二、固相萃取方法的建立与优化
固相萃取技术使用起来虽然比液液萃取简单,但建立一个固相萃取的方法并无快捷方式可走。
建立固相萃取方法必须考虑与萃取过程相关的多种因素,归纳起来可通过下图来了解:
方法建立如下图片1.jpg:
方法建立如下图片2.jpg:
1、初步固相萃取方法的建立
建立初步的萃取方法要考虑:
·
选择合适的SPE柱
选择合适的固相萃取方法
方法的优化
2、固相萃取柱的选择<
1)柱填料的选择
首选根据目标化合物与干扰物的差异,如极性,分子量,pka值等,选择合适的填料。
固相萃取柱的选择如下图片.jpg:
2)固相萃取柱规格的选择对于反相、正相和吸附型固相萃取柱来说,被萃取样品的质量不超SPE柱填料的5%(参考值,同一种SPE柱对不同的目标物选择性不同,吸附容量不同);
<
P0cm="
0cm"
0cm?
0pt?
>
离子交换型的固相萃取柱,必须考虑离子交换的容量。
不同厂家的小柱离子交换容量稍有差异。
下表附SPE小柱的容量和洗脱参数
SPE柱上样容量和洗脱体积的选择
规格
最大上样量
最小洗脱体积
100mg/1mL
5mg
250µ
L
200mg/3mL
10mg
500µ
500mg/6mL
25mg
1.2mL
1g/6mL
50mg
2.4mL
3、选择合适的固相萃取方法
固相萃取的保留机制可分为两种:
吸附剂(填料)保留目标化合物:
绝大多数化合物应用此机制,填料保留其目标组分及少量杂质,通过淋洗步骤去除吸附在柱上的少量杂质,最后选择合适的(洗脱)溶剂把目标组分洗脱下来。
根据吸附剂的保留机理可进一步分为:
(1)反相(C18,C8,CN,Phenyl,C4,C1)
分析物:
非极性至中等极性
基质:
水溶性
方法:
a.活化:
通常用水溶性有机溶剂如甲醇活化,然后用水平衡
b.淋洗:
含0-50%极性溶剂的缓冲溶液淋洗杂质
c.洗脱:
极性或非极性溶剂洗脱目标物
(2)正相(Silica,Florisil,Diol,NH2)
分析物:
中等极性到强极性
基质:
方法:
非极性有机溶剂b.洗脱:
非极性有机溶剂
如下图片:
(3)阳离子交换(SCX,PRS,COOH)
u
阳离子(碱性)化合物
1.活化:
用于非极性有机溶剂中的样品时,可用样品溶剂来活化;
在用于极性溶剂中的样品时,可用水溶性有机溶剂过柱后,然后用水平衡,最后再用适当pH值的缓冲溶液进行平衡。
2.上样:
样品溶液pH值要小于其pKa两个单位(以保证其带电荷)
3.洗脱:
洗脱溶液pH值要大于其pKa两个单位(中和分析物的电荷)
(4)阴离子交换(SAX,PSA,NH2,PAX/MAX)
阴离子(酸性)化合物
在用于极性溶剂中的样品时,可用水溶性有机溶剂活化后,然后用水平衡,最后再用适当pH值的缓冲溶液进行平衡。
样品溶液pH值要大于其pKa两个单位(以保证其带电荷)。
洗脱溶液pH值要小于其pKa两个单位(中和分析物的电荷)。
吸附剂(填料)保留杂质:
食品中色素等杂质的去除多用此机制。
填料保留杂质而不保留或只保留极少量的目标组分,所以上样后即开始收集目标组分,最后用目标物所在的溶剂进一步洗脱。
合并两部分收集液。
(1)活化:
以样品所在的有机溶剂进化活化,1-2柱管体积
(2)上样:
提取液转移至柱内,并收集流出液
(3)洗脱:
用样品所在的有机溶剂进一步洗脱,收集流出液。
合并上样和洗脱流出液。
4、固相萃取方法的优化
1)影响萃取效率的因素
(1)填料(固定相)-----核心
选择合适的SPE柱是保证理想结果的前提。
(2)洗脱溶剂的强度:
采用正相固定相时,溶剂强度随其极性增强而增加;
采用反相固定相时,溶剂强度随极性减弱而增强。
(3)pH值:
离子交换固定相、被分析物和干扰物质的pKa各不相同。
通过调节pH大小,可以使固定相带电荷,被分析物带相反电荷,而使干扰物质不带电荷;
或者反过来,使固定相带电荷,干扰物质带相反电荷,而使被分析物不带电荷。
(4)操作:
控制合适的流速、活化的时不要让柱干涸等
2)常见问题及解决方法
分析物回收率低
萃取重现性差
洗脱馏分中含有干扰物
SPE柱流速降低或阻塞
具体解决方案如下:
A.
•
未保留?
被淋洗?
未被洗脱或部分洗脱?
首先要把上样液、淋洗液、洗脱液均收集,进样分析,确定问题来源
回收率差如下图片:
重现性差如下图片:
相关图片如下:
相关图片如下:
举例说明1.
参考文献方法用C18柱做相关药物的净化,过柱方法如下:
分别用乙酸乙酯、甲醇和水活化小柱,然后把处理过的样品过柱(溶剂为具一定pH值得缓冲溶液),水淋洗小柱后,用乙酸乙酯洗脱目标物。
结果:
接受液混浊状,回收率和重现性都不理想,可能是什么原因呢?
答案:
正确的做法是要在淋洗过程结束后把小柱完全抽干。
原因有二,其一因为淋洗溶剂(水)与洗脱溶剂(乙酸乙酯)不互溶,如果不抽干洗脱溶剂与目标物不能充分作用,所以造成回收率和重现性都没有保证,同时从外观上看接受液是液混浊液;
其二如果淋洗过程不抽干小柱,洗脱溶剂里会引入水(淋洗剂),对下一步浓缩造成很大的困难。
2、水中的灭草松前处理方法
取500ml水样过滤,待过CleanertPEP柱(相当于WatersHLB)净化
1)
用5mL四氢呋喃洗柱子,除掉杂质
2)
用5mL甲醇1mL/min活化柱子
3)
用5mL纯水1mL/min活化柱
4)
500mL的水样以5mL/min的速度过柱
5)
5mL纯水2mL/min淋洗
6)
小柱真空抽干20min
7)
0.9mL的甲醇1mL/min淋洗,弃去淋洗液
8)
3mL四氢呋喃1mL/min的速度洗脱柱子,收集洗脱液浓缩定容至3mL液相检测。
回收率不理想,请问此方法有何问题?
因为目标物灭草松呈酸性,pKa=3.3,而选择的小柱PEP是极性的。
所以正确的做法是样品在过柱之前一定要调节水样的pH值小于等于3,使目标物分子化,以保证目标物能被小柱充分保留,否则在上柱的过程中容易造成“漏”的现象,从而造成回收率差。
三、固相技术应用实例解析
1、食品领域应用
1)动物组织中盐酸克仑特罗等4种β-激动剂药物残留检测(CleanertPCX,P/N:
CX1506)
1.实验材料
1.1固相萃取小柱:
PCX(150mg/6mL)
1.2四种β-激动剂药物:
盐酸克仑特罗、沙丁胺醇、西马特罗、莱克多巴胺等4种β-激动剂药物。
2.试样的制备
取猪肝空白样品,经过液液萃取初步处理后,添加适宜浓度的标准溶液作为空白添加试样。
3.净化
依次用甲醇5mL、水5mL和30mmol/L盐酸5mL润洗固相萃取小柱,将上述备用液过柱,依次用水5mL、甲醇5mL淋洗,真空抽干,用4%氨化甲醇5mL洗脱PCX小柱,收集洗脱液于具塞玻璃试管中,50℃下氮气吹干。
在样液过柱和洗脱过程中流速控制在1mL/min左右。
4.衍生化及检测
将上述盛有残渣的具塞玻璃试管放入50℃烘箱中加热片刻,除去水分后,加入甲苯100mL和双三甲基硅基三氟乙酰胺(BSTFA)100mL,涡旋振荡20s,密封玻璃塞,置于80℃恒温烘箱中加热1小时,冷却后加入300mL甲苯,作为试样溶液,供气相色谱-质谱分析(色谱柱:
DA-5MS,30m×
0.25mm×
0.25µ
m,P/N:
1525-3002)。
5.结果
5.1回收率实验(精密度和准确度)
将猪肝空白样品经过液液萃取初步处理后,分别添加一定量的标准溶液,配制1μg/L、2μg/L、5μg/L、10μg/L和100μg/L五个浓度的试样溶液,每批次内同一浓度做5次平行实验,共4个批次(样品典型回收率色谱图见附图)。
猪肝中实验结果列表如下
添加浓度
(μg/L)
回收浓度
平均回收值
平均回收率
(%)
相对标准偏差
1
0.75
0.72
72.40
5.93
0.67
0.70
0.78
2
1.62
1.63
81.30
1.23
1.66
1.60
1.61
1.64
5
4.02
4.24
84.80
4.16
4.10
4.27
4.38
4.43
10
8.24
8.45
84.45
2.81
8.35
8.77
8.62
8.25
100
90.24
9.12
91.15
2.86
87.15
91.77
92.62
93.95
5.
 升级会员
升级会员 