肿瘤新十大特征.docx
《肿瘤新十大特征.docx》由会员分享,可在线阅读,更多相关《肿瘤新十大特征.docx(22页珍藏版)》请在冰豆网上搜索。
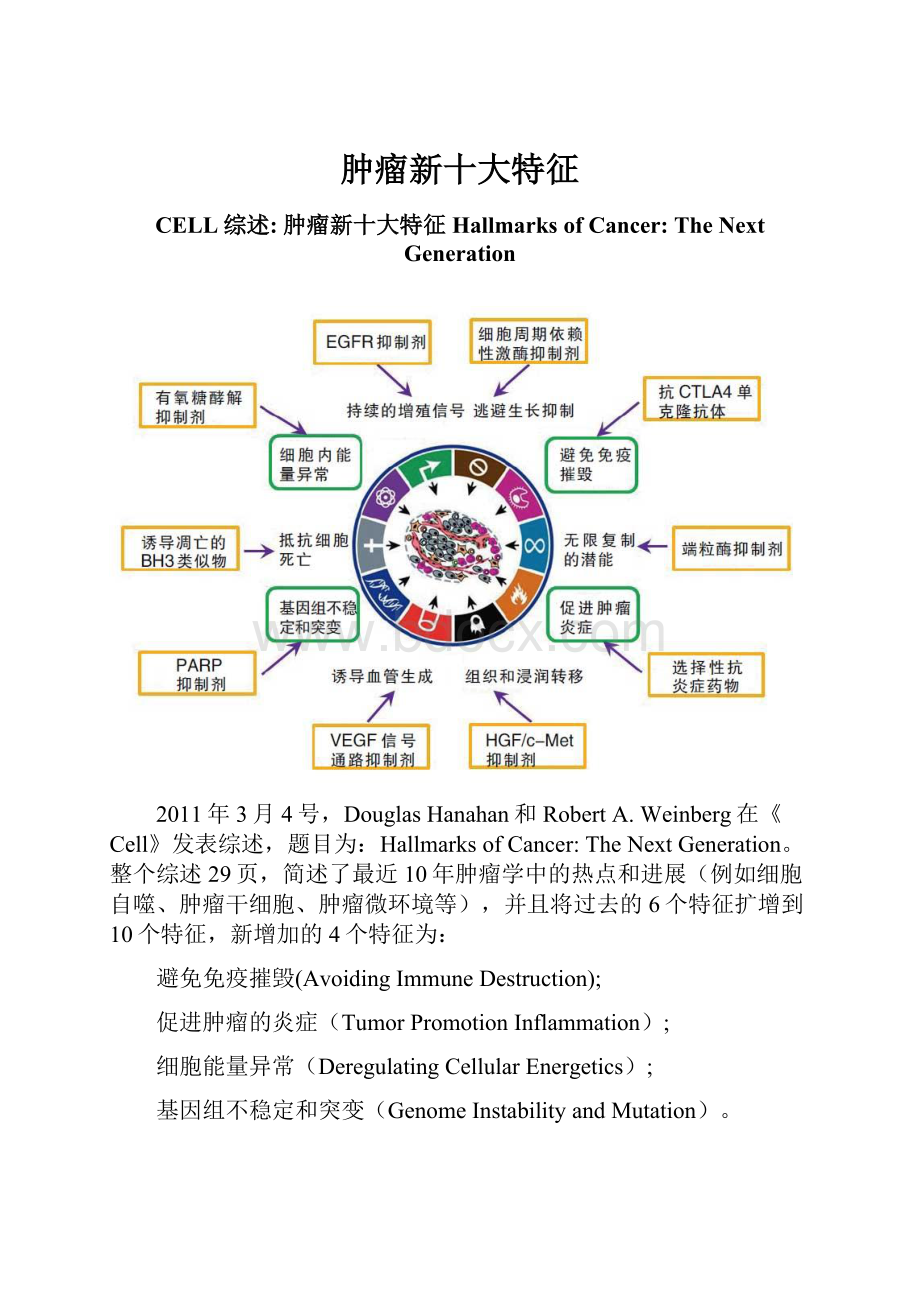
肿瘤新十大特征
CELL综述:
肿瘤新十大特征HallmarksofCancer:
TheNextGeneration
2011年3月4号,DouglasHanahan和RobertA.Weinberg在《Cell》发表综述,题目为:
HallmarksofCancer:
TheNextGeneration。
整个综述29页,简述了最近10年肿瘤学中的热点和进展(例如细胞自噬、肿瘤干细胞、肿瘤微环境等),并且将过去的6个特征扩增到10个特征,新增加的4个特征为:
避免免疫摧毁(AvoidingImmuneDestruction);
促进肿瘤的炎症(TumorPromotionInflammation);
细胞能量异常(DeregulatingCellularEnergetics);
基因组不稳定和突变(GenomeInstabilityandMutation)。
并且将过去的回避凋亡(EvadingApoptosis),调整为抵抗细胞死亡(ResistingCellDeath)。
背景介绍
我们已经提出了总共6种肿瘤标志,组成了一个基本原理,它提供了一个理解肿瘤性疾病显著差异的逻辑网络(HanahanandWeinberg,2000)。
我们的讨论中包含了这样的概念,肿瘤细胞逐渐进展成新生物,它们获得一系列标志性能力,而人类肿瘤形成的多步骤的过程可以用初始癌细胞获得使它们成为肿瘤并最终表现出恶性肿瘤的特征。
我们注意到由于附属结构的存在,肿瘤不只是癌细胞组成的岛状物。
它们是由多种不同类型的细胞组成的复合物,这些细胞之间存在异质的相互作用。
我们是这样描述招募来的正常细胞,它们以主要参与者的身份形成肿瘤相关基质,而不是作为旁观者;这样的话,这些间质细胞对于特定能力标志的发展和表达是有贡献的。
在接下来的十年中,这个概念已经得到确认和发展,就是肿瘤生物学不能只是通过对这些癌细胞特性描述来进行简单的理解,而是应该包括“肿瘤微环境”对肿瘤形成的贡献。
直到本文出版为止,癌症研究的非凡进程中,新的研究致力于阐明和订正标志能力的初始形式。
另外也有部份研究提出了我们在初始标志特征中未完整的问题和突出性机制概念。
受这些进展的鞭策,我们重新回顾了初始标志,考虑到新的概念应该包含到这个名单中,并对招募来的基质细胞的功能及其对肿瘤生物学的贡献进一步阐述。
标志能力—概念上的进展
癌症的6个标志-这些特征性而且互补的能力使肿瘤可以生长并向远处转移,持续地为理解癌症生物学提供坚实的基础。
(图1;见当前图片的可下载补充信息)。
在这篇综述的第一部份,我们简要地回顾了2000年初始描述的各个标志的本质,并附上选择性的概念性进展的插图(用副标题的斜位字区别),这些是过去10年来的概念进展,可以用于理解它们的基本机制。
接下来的部份,我们阐述了拓宽概念深度的新进展,按顺序描述了导致获得这6种标志性能力决定性特征,两种新发现的标志性能力,肿瘤微环境的结构和信号相互作用,这两种能力决定了癌症的表型,最后我们讨论这些概念在治疗应用方面的新领域。
维持增殖的信号
癌细胞最值得争论的基本特征之一是它们持续慢性增殖的能力。
正常组织小心地控制着生长启动信号的产生和释放,这些信号指导进入细胞增殖和分化周期并在这个周期运行,从而维持细胞数量的稳态,从而维持正常组织结构结构和功能。
癌细胞通过下调这些信号而变成它们自已命运的主宰。
这些启动信号大多数是由结合在细胞表面受体上的生长因子来传输的,典型特征是包含了细胞内酪氨酸激酶区域。
这个区域通过胞内信号途径发送信号,从而调节细胞通过细胞周期,表现为细胞增殖(也就是细胞数量的增加);通常这些信号也影响细胞生物学性质,如细胞生存和能量代谢。
值得注意的是,精确地鉴定和寻找在正常组织中开启的增殖信号源头在上一个十年中是很难理解的,而现在仍然如此。
而且我们对于这个控制丝裂原信号释放的机制仍然知之甚少。
对于这个机制的部份理解是与以下事实并存的,细胞因子的信号控制细胞的数量和在组织中的位置,是通过一个短暂的而且是空间调节的方式从一个细胞向它的周围细胞传递;这种旁分泌信号在实验中难以评价。
另外生长因子的生物利用度的调节通过以下方式进行:
隔离细胞周围空间和细胞基质,活化蛋白酶、硫酸酯酶以及其它对这些酶类的释放和活化有关的网络,显然这是一个高度特异性和局限性的方式。
相对来说,癌细胞中的丝裂原信号是比较好理解的(LemmonandSchlessinger,2010;Witschetal.,2010;HynesandMacDonald,2009;Perona,2006)。
癌细胞可以通过多种代替途径获得持续增殖的信号:
它们可能自已产生生长因子配体,从而通过表达同源受体作出反应,结果导致自分泌增殖剌激。
另一种替化方法是,癌细胞可能发送信号到肿瘤相关支持间质中的细胞,作为后者的反应提供了各种生长因子给癌细胞(Chengetal.,2008;Bhowmicketal.,2004).受体信号也可以通过上升表达于癌细胞表面的受体蛋白水平而下调,对数量有限的配体生长信号产生高度的反应性表现。
同样的结果出现在受体分子结构改变中,使配体依赖的启动更为方便。
生长因子依赖可能是从这些受体下游信号启动途径复合物结构活化衍生而来,避免了剌激这些配体介导的受体活途径的需求。
由于许多下游信号途径是由配体剌激的受体发出的,如来自Ras信号传感器的反应,可能只是一个由活化受体传导亚类的代表。
体细胞突变活化依赖的下游途径
癌细胞基因组的高通量DNA测序显示了在某些人类肿瘤中存在体细胞突变,这预示着通常由活化的生长因子受体激活的信号回路的基本激活。
我们目前已经明确的是40%人类恶性黑色素瘤包含了活化的变异,可以影响B-Raf蛋白,从而通过Raf到丝裂原活化蛋白(MAP)激酶途径(DaviesandSamuels 2010)。
相似地,在大量的肿瘤类型中检测出PI3-K催化亚单位的亚型,它们服务于高度活化的PI3-K回路,包括关键性的Akt/PKB信号转导蛋白(JiangandLiu,2009;YuanandCantley,2008)。
在生长因子受体发出的多种途径相互作用的功能影响方面,活化上游(受体)与下游(转导蛋白)信号对肿瘤的作用仍不明确。
破坏减弱增殖信号机制的负反馈
最近的研究结果突出了负反馈链的重要作用,正常情况下它的启动是用于减缓各种类型的信号,从而通过细胞内回路的信号流得到稳定的调节。
(WertzandDixit,2010;CabritaandChristofori,2008;Amitetal.,2007;Mosessonetal.,2008).这些负反馈机制的缺点在于具有增强增殖信号的功能。
这一类型调节的原型包括Ras癌蛋白:
Ras的致癌效应并不是它本身信号活性高度活化的结果;而是致癌变异影响了ras基因而减弱了GTP酶活性,它作为一个本质上是负反馈的机制启动,这个机制平时是用于确保活化的信号传递是短暂的。
相似的负反馈机制在增殖信号回路中的多个节点中启动。
一个重要的例子是PTEN磷酸化酶,它通过降解PIP3而抵消PI3K的功能。
PTEN的无功能变异放大了PI3K的信号并启动多种癌症实验模型的肿瘤生成;在人类肿瘤中,PTEN的表达通常是通过启动子甲基化完成的(JiangandLiu,2009;YuanandCantley,2008).
另一个例子是mTOR激酶,一个细胞生长和代谢的协调子同时位于PI3K途径的上下游。
在某些癌细胞内的回路中,mTOR的活化通过负反馈导致PI3K信号的抑制。
这样的话,当癌细胞内的mTOR在药理学水平上(如雷帕霉素)被抑制,相关负反馈的丢失导致PI3K和它的效应子Akt/PKB活性的增加,进而减弱mTOR的抗增殖抑制效应(SudarsanamandJohnson,2010;O’Reillyetal.,2006).人类癌细胞中可能将证明广泛存在一种或另一种的信号途径减弱负反馈环,作为一种使细胞达到非依赖性增殖的方式。
此外,干扰这种减弱信号可能有利于对有丝分裂信号靶向治疗药物的适应性抵抗的进展。
过度的增殖信号能触发细胞衰老
早期研究癌基因的作用确定了这样一个概念,这些基因的不断表达以及已经证明的这个信号所致的蛋白产物导致相应的癌细胞增殖速度增加从而肿瘤生长。
最近的研究破坏了这个概念,由RAS,MYC和RAF等癌蛋白剌激的信号过度表达可以引起来自细胞的抵抗反应,特别是诱导细胞衰老或/和死亡(ColladoandSerrano,2010;Evanandd’AddadiFagagna,2009;Loweetal.,2004)。
例如,培养的细胞表达高水平的Ras癌蛋白可能进入非增殖但能存活的状态,称为衰老;相反的,如果细胞表达低水平Ras可能避免衰老而进入增殖。
衰老细胞具备的特征,包括增大的细胞质,缺乏增殖标志,以及表达衰老诱导的β-半乳糖酶,这些特征在经设计的过度表达特定癌基因的小鼠组织中大量出现。
(ColladoandSerrano,2010;Evanandd’AddadiFagagna,2009)并且在一些人类黑色素瘤病例中普遍存在(MooiandPeeper,2006)。
这些相互矛盾的表现反应了不同的细胞防御机制,以减弱细胞经历过高水平的特定类型信号。
相应地,癌细胞内的致癌信号可能表现为在最大剌激有丝分裂和避免这些抗增殖防御之间的妥协。
作为代替方案,一些癌细胞可能通过使这些衰老或凋亡诱导回路失去功能而适应高水平的致癌信号。
负性调节细胞增殖
逃避生长抑制除了诱导和持续的正性剌激生长信号的标志性能力外,癌细胞应该也具备有力的方案以负性调节细胞增殖;这些方案中的大部分依赖于抑癌基因。
成打的肿瘤抑制子可以在不同途径上限制细胞的生长和增殖,这些是通过这种或那种方式对动物或人类癌症进行特征性抑制发现的;很多这些基因已经通过小鼠实验以获得或失去功能来确实真正是肿瘤抑制子。
两种典型的肿瘤抑制子编码RB(视网膜母细胞相关)和TP53蛋白;它们在两个关键的细胞调节互补回路中起着中央控制节点的作用,控制着决定细胞增殖或代替以活化衰老和凋亡程序。
RB蛋白整合来自胞内和胞外的不同信号,并相应地,决定细胞是否通过细胞生长和分裂周期(BurkhartandSage,2008;Deshpandeetal.,2005;SherrandMcCormick,2002).RB途径存在功能性缺陷的癌细胞缺乏了细胞周期进程的关键性守门员的功能,它的缺席允许细胞持续增殖。
由于RB转导来自细胞外的大部份生长抑制信号,TP53接受来自功能区在胞内的操作系统的压力和不正常感受器的输入信号:
如果基因组损伤的级别过度,或如果核苷酸库水平,生长启动信号,乳糖,或氧合水平不在最佳状态,TP53将使细胞周期进程停止直至这个状态正常化。
另一种代替方法,当这个细胞亚系统的警戒信号到了无可挽回或损伤无法弥补时,TP53将触发凋亡。
值得注意的是,活化的TP53的效应是复杂的而且是环境依赖性的,依细胞类型以及细胞压力严重性和压力抵抗情况,还有基因组损伤而变。
虽然两种经典的增殖抑制-TP53和RB-具有重要的调节细胞增殖的功能,多个系列的证据表明它们各自作为一个更大的网络的部份,这些网络作为一个功能冗余。
例如,嵌合体小鼠全身普遍都具有Rb基因缺乏的细胞,但令人惊讶的是没有出现细胞增殖异常,除了本来期望Rb功能的缺失将允许这些细胞以及它们传代细胞的分化周期持续进行;一些Rb缺乏细胞株将令人满意地进展成肿瘤。
同时,在这些嵌合体小鼠体内的缺乏Rb细胞参与了相对正常组织形态;唯一成为肿瘤的是晚期垂体中(LipinskiandJacks,1999)。
相似地,TP53缺乏的小鼠发育正常,表现为大量的正常细胞和组织稳态,而在生命的晚期再次发展成不正常,表现成白血病和肉瘤(GhebraniousandDonehower,1998)。
以上两个例子应该可以说明导致缺乏这些关键性增殖抑制子细胞持续不正常复制的启动和冗途机制。
接触抑制机制和肿瘤的逃避
40年来的研究证明二维培养中密集的正常细胞群体形成细胞与细胞之间的接触,产生汇合的细胞单层。
重要的是,这种“接触抑制”在各种培养的癌细胞中被废除了,这表明接触抑制是体内启动的用于确保正常组织稳态机制在体外的替代,这是在致癌过程中被废除的。
直到最近,生长控制的机制基础仍不明确。
然而,接触抑制的机制开始出现。
机制之一包含了NF2基因的产物,一直作为肿瘤抑制基因,因为它的缺失触发了人类成纤维瘤病的一种形式。
Merlin蛋白,NF2的胞质产物,通过成对的细胞表面粘附分子(如E-cadherin)到跨膜酪氨酸激酶(如EGF受体)形成接触抑制。
以这种方式,Merlin蛋白加强了钙粘蛋白介导的细胞与细胞间的粘附性。
另外,通过分离生长因子受体,Merlin蛋白限制它们有效地发射丝裂原信号的能力(Curtoetal.,2007;Okadaetal.,2005)。
第二种接触抑制的机制包括LKB1表皮极性蛋白,它们可以组织表皮结构并帮助维持组织完整性。
例如,当组织中Myc癌基因上调时,LKB1能管理强大的Myc癌基因的促有丝分裂效应使表皮结构处于静息状态;相反,当LKB1的表达受抑制时,表皮完整性变得不稳定,而表皮细胞对Myc诱导的转化变得更敏感(Partanenetal.,2009;HezelandBardeesy,2008)。
LKB1同时已经确定是一个抑癌基因,在某些人类肿瘤中缺失(Shaw,2009),这可能反应它的正常功能是作为不正当增殖的抑制子。
人类癌症中这两种接触介导的生长抑制缺失的频率仍需要继续研究;毫无疑问的是其它由接触介导的可以阻止增殖的机制仍需经进一步研究确认。
这些清楚的机制使细胞可以构建和维持组织结构的复杂性,成为对不正常增殖信号的抑制和平衡的重要方式。
TGF-b最出名的是它的抗增殖效应,而目前人们更仔细研究癌细胞如何逃避这些效应而较少研究简单地关闭这些信号回路(Ikush-imaand Miyazono, 2010; Massague,2008; Bierieand Moses, 2006)。
在很多晚期肿瘤,TGF-β信号已经偏离细胞增殖抑制,而是用于激活一种细胞程序,称为表皮到间质的转换(EMT),这使细胞的特征与高度恶性相关,相关细节将在下文阐述。
抵抗细胞死亡
在最近的20年,细胞凋亡这一概念,通过程序性细胞死亡充当癌症发展的自然屏障,已经建立了进行令人信服的功能性研究,(AdamsandCory,2007;Loweetal.,2004:
EvanandLittlewood,1998).
对控制着凋亡程序信号回路的阐释揭示了凋亡在不同的生理压力下是如何被触发的,这些压力是癌细胞在肿瘤形成过程中遇到或者是抗癌治疗的结果。
在凋亡诱导的压力中值得注意的是由于癌基因信号的提高导致的信号不平衡,就像早先所提及的那样,并且DNA损伤与高增殖相关。
同时其他研究显示在成功进展到高度恶性和对治疗抵抗的肿瘤中凋亡是减弱的。
(AdamsandCory,2007;Loweetal.,2004)。
凋亡机制是由上游调节子和下游效应子共同构成的(AdamsandCory,2007)。
按顺序,调节子分成两个主要的回路,一个是接受和传递胞外死亡诱导信号式(外部的凋亡程序,包括例如Fas配体/Fas受体),另外一个感知和综合一系列胞内起源的信号(内在程序)。
它们每个最终都激活一个正常的潜在蛋白酶(分别是半胱氨酸天冬氨酸蛋白酶8和9),从而开始一个级联的蛋白水解,包括凋亡执行阶段效应蛋白酶,这个阶段细胞渐渐分解后消失,这个效应由它的邻近细胞和专门的吞噬细胞完成。
目前,内在凋亡程序更广泛地作为癌症致病的屏障。
在调节子和效应子之间传递信号的“凋亡扳机”是通过平衡促进和抵抗凋亡的Bcl-2家族成员的调节蛋白来完成的(AdamsandCory,2007)。
原型是Bcl-2,连同它最近的家族成员(Bcl-xL,Bcl-w,Mcl-1,A1)都是凋亡抑制子,通过与两个前凋亡触发蛋白(BaxandBak)结合并起抑制作用在多数场合起作用;后者嵌在线粒体外膜中。
当受到它们抗凋亡家族成员抑制减弱时,Bax和Bak破坏线粒体外膜的完整性,从而导致前凋亡信号蛋白的释放,它们中最主要的是细胞色素C。
细胞色素C依次激活蛋白酶级联,通过它们的蛋白水解酶活性产生与凋亡程序相关的多种细胞改变。
Bax和Bak与抗凋亡的Bcl-2样蛋白共用蛋白-蛋白相互作用区,称为BH3基序,从而介导它们的各种物理相互作用。
相关蛋白(每个蛋白包含单个像BH3的基序)的亚家族与各种细胞感受器配对而异常活化;这些“只有BH-3”的蛋白通过干扰抗凋亡Bcl-2蛋白或通过直接剌激这个家族的凋亡前成员而活化(AdamsandCory,2007;WillisandAdams,2005).虽然触发凋亡的细胞状态仍没有完成列举,但是多个在肿瘤进展中起关键作用的异常感受器已经明确(AdamsandCory,2007;Loweetal.,2004)。
最值得注意的是通过抑癌基因TP53起作用的DNA损伤感受器(JunttilaandEvan,2009);TP53通过上调Noxa和Puma这两个只有单个BH3基序蛋白的表达诱导凋亡,从而相应地导致大量的DNA断裂和其它染色体的不正常。
另一种替代方式是,不充分的生长因子信号(如淋巴细胞上的白细胞介素-3或上皮细胞上胰岛素样的生长因子1/2数量的不充足)能通过称为Bim的单个BH3基序蛋白诱导凋亡。
导致细胞死亡的另一个情况也包括某些癌蛋白的过度活化,如Myc,它除了被抗凋亡信号平衡的情况外可以触发凋亡(部分通过Bim和其它单个BH3基序蛋白)(JunttilaandEvan,2009;Loweetal.,2004).肿瘤细胞发展出一系列可以限制或防止凋亡的类型。
最普遍的是抑癌基因TP53功能的缺失,这个类型缺失了来自诱导凋亡回路的重要感受器。
存在的一些代替的方式如下,肿瘤可能获得相似的结果,这是通过增加抗凋亡调节子(Bcl-2,Bcl-xL)或生存信号(Igf1/2),或下调凋亡前因子(Bax,Bim,Puma)或缩短胞外配体诱导的死亡途径。
避免凋亡的机制的多样性反应了癌细胞株在发展成恶性状态过程中遇到的凋亡诱导信号的多样性。
在最近的十年,凋亡机制和程序结构以及癌细胞逃避凋亡作用的分类得到广泛关注。
从那以后,值得关注的概念进展就包括了其他方式的细胞死亡,这就拓宽了“程序化细胞死亡”的领域成为一种阻碍细胞癌变的屏障。
自吞噬介导了癌细胞生存和死亡
自吞噬与凋亡一样成为一种重要的细胞生理反应,正常细胞中的启动处于低的基础的水平,但在特定的细胞压力状态下能被强烈地诱导,最明显的压力状态是营养缺乏(Levine andKroemer, 2008; Mizushima, 2007). 自吞噬程序使细胞内的细胞器(如核糖体和线粒体)被破坏,使分解代谢产物用于生物合成和能量代谢从而得到循环。
当降解发生时,作为这个程序的一部份,胞内囊泡称为自吞噬体包裹胞内细胞器并把它们和溶酶体混和。
以这种方式,低分子量的代谢的产物支持了多数癌细胞在压力和营养受限的环境中生存。
与凋亡相似,自吞噬机制也有调节子和效应子复合物(Levineand Kroemer,2008; Mizushima,2007)。
后者是一种介导自吞噬体的形成并把它传送到溶酶体的蛋白质。
令人关注的是,最近的研究显示控制自吞噬,凋亡和细胞稳态三个回路之间的存在交叉。
例如,包括PI3-kinase,AKT,和mTOR激酶的信号途径由生存信号剌激以阻止凋亡,同时也抑制自吞噬;当生存信号不足时,PI3K信号途径会被下调,从而导致自吞噬和/或凋亡的减少(Levineand Kroemer,2008;Sinha and Levine,2008;Mathew etal.,2007)。
另一个这两种程序的联系存在于Beclin-1蛋白,这个蛋白已经通过遗传学研究证实是诱导自吞噬所必需的(LevineandKroemer,2008; Sinha and Levine, 2008; Mizushima, 2007)。
Beclin-1是凋亡调节蛋白BH3-only亚家族成员之一,它的BH3区域允许它与Bcl-2/Bcl-xL蛋白结合。
压力感受器和BH3蛋白配对后能从Bcl-2/Bcl-xL替换下Beclin-1,释放的Beclin-1能触发自吞噬,就像它们释放Bax和Bak触发凋亡一样。
因而压力转导的BH3蛋白(如,Bid、Bad、Puma等)可以减少依赖于细胞生理状态的凋亡和/或自吞噬。
带有失活的Beclin-1等位基因或特定的其它自吞噬机制复合物的小鼠的癌症易感性增加(Whiteand DiPaola, 2009:
LevineandKroemer,2008)。
这个结果提示诱导自吞噬可以作为一个致癌的屏障,它的启动不依赖于凋亡甚至可以说与凋亡不一致。
相应地,自吞噬明显地也可以是另一个屏障,这是肿瘤进展时必需绕过的(Whiteand DiPaola,2009)。
可能矛盾的是,对癌细胞的营养缺乏、放射治疗和特定的细胞毒药物能引起自吞噬水平的增高,这对癌细胞明显是一种细胞保护,只是损伤而不是这些压力诱导条件所致的杀伤(Whiteand DiPaola, 2009;Apeletal.,2009;Amaravadi and Thompson,2007; Mathew et al.,2007)。
此外,处理严重压力条件下的癌细胞可以通过自吞噬而缩小并进入一个可逆的休眠状态(WhiteandDiPaola, 2009; Lu et al., 2008)。
这种生存反应可能使一些晚期肿瘤在有效的抗肿瘤药物治疗后能残留并最终再长出来。
因此,与TGF-β信号相似,在肿瘤形成的早期阶段起抑制作用而在晚期起促进作用,自吞噬像是对肿瘤细胞有着相反的作用,从而肿瘤能得到进展(Apeletal.,2009; WhiteandDiPaola, 2009)。
将来研究中的重要日程必需包括澄清遗传学和细胞生理状态,这些能展示自吞噬在什么时间以及如何使癌细胞生存或导致它们死亡。
坏死具有促进炎症和肿瘤的潜能
与凋亡相反,凋亡中死亡的细胞变成一个几乎看不见的尸体并立即被周围细胞消耗光,而坏死细胞会肿胀破裂,并释放出它们的内容物进入其周围微环境。
以往认为坏死更像是有机死亡,是一种系统广泛地耗尽和崩溃,这个概念的全貌已经发生了改变:
细胞的坏死在某些情况下明确是在基因控制之下,而不是随机的无序的进程(Galluzzi andKroemer,2008;Zongand Thompson,2006)。
或者更重要的是,坏死细胞死亡后释放促炎症信号进入周围组织微环境,这与没有这种现象的凋亡和自吞噬相反。
结果,坏死细胞能招募免疫系统的炎症细胞。
(Grivennikov et al., 2010; White et al.,2010; Galluzziand Kroemer,2008),炎症细胞的作用是专门监督组织损伤的程度并移除相关的坏死残骸。
然而在肿瘤背景下,多方面的证据显示免疫炎症细胞可能是活跃的肿瘤促进因素,因于这个原因,这些细胞具有促进血管生成、癌细胞增殖和侵袭功能(如下)。
另外,坏死细胞能释放生物活性调节因子,如IL-
 升级会员
升级会员 