脱硫化学分析.docx
《脱硫化学分析.docx》由会员分享,可在线阅读,更多相关《脱硫化学分析.docx(42页珍藏版)》请在冰豆网上搜索。
脱硫化学分析
烟气脱硫石膏化学分析
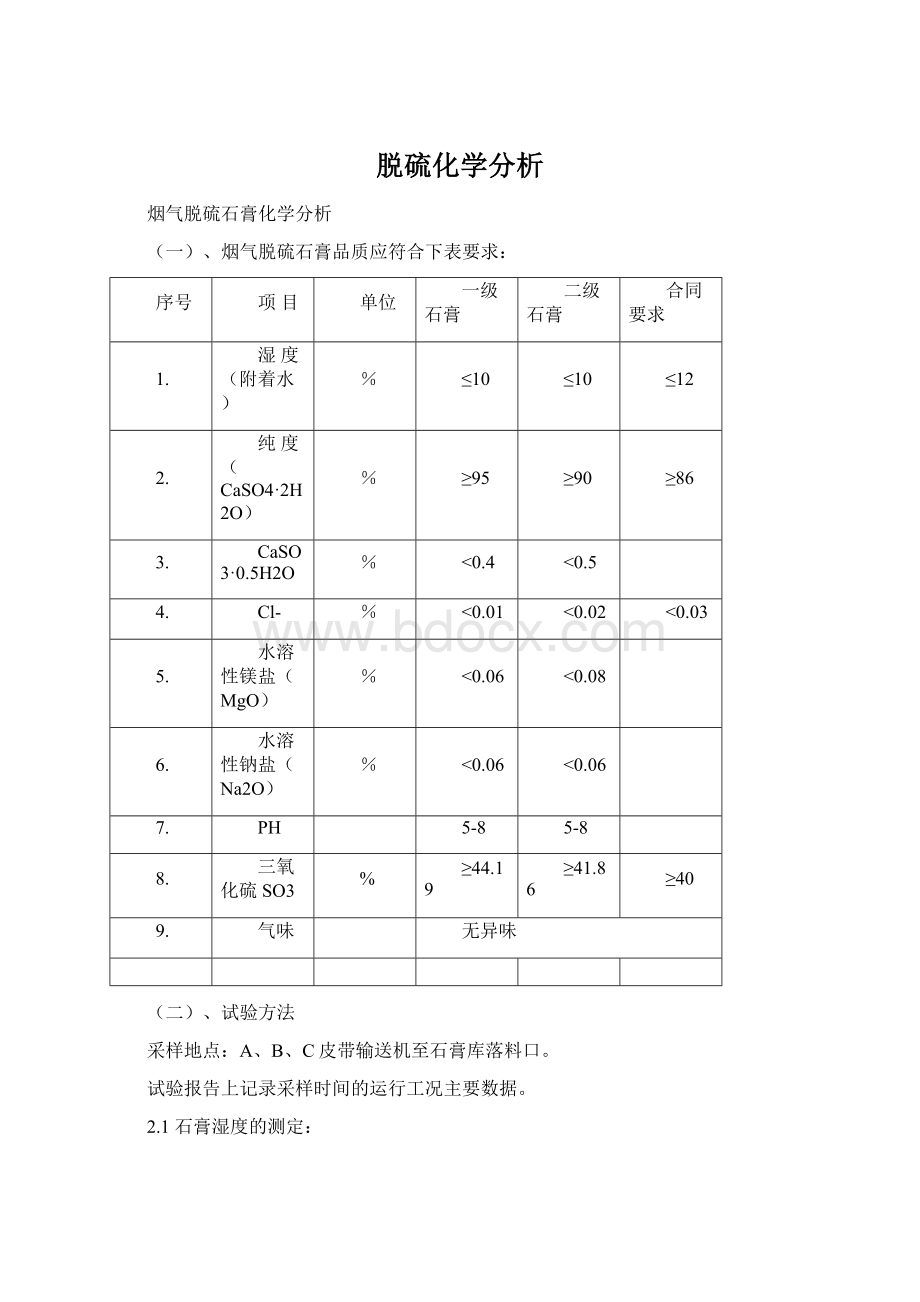
(一)、烟气脱硫石膏品质应符合下表要求:
序号
项目
单位
一级石膏
二级石膏
合同要求
1.
湿度(附着水)
%
≤10
≤10
≤12
2.
纯度(CaSO4·2H2O)
%
≥95
≥90
≥86
3.
CaSO3·0.5H2O
%
<0.4
<0.5
4.
Cl-
%
<0.01
<0.02
<0.03
5.
水溶性镁盐(MgO)
%
<0.06
<0.08
6.
水溶性钠盐(Na2O)
%
<0.06
<0.06
7.
PH
5-8
5-8
8.
三氧化硫SO3
%
≥44.19
≥41.86
≥40
9.
气味
无异味
(二)、试验方法
采样地点:
A、B、C皮带输送机至石膏库落料口。
试验报告上记录采样时间的运行工况主要数据。
2.1石膏湿度的测定:
取一称量瓶称其重量A(mg),加入约10g的湿石膏,称取称量瓶和石膏的总重量B(mg)。
将称量瓶放入烘箱中在45℃下干燥1个小时至恒重,再次烘干0.5小时,两次误差不超过0.02g。
称取干燥后称量瓶和干石膏的总重量C(mg)。
计算公式如下:
2.2结晶水的测定(通过测定结晶水计算石膏中CaSO4·2H2O含量)
称取约1g干石膏试样(M1),精确至0.0001g,放入已烘干、恒重的带有磨口塞的称量瓶中,于230±5℃的烘箱中加热1h,用坩埚钳将称量瓶取出,盖上磨口塞,放入干燥器中冷至室温,称量。
再放入烘箱中于同样温度下加热30min,如此反复加热、冷却、称量至恒量(M2)。
X2=4.7778×100×(M1-M2)/M1
式中:
X2――石膏的纯度,%
M1――加热前试料的质量,g
M2――加热后试料的质量,g
2.3石膏中CaSO3·0.5H2O含量的测定
方法原理:
在酸性条件下,样品中的亚硫酸盐与碘进行氧化还原反应,过量的碘以硫代硫酸钠标准溶液进行滴定。
该试验涉及的化学反应式为:
SO32ˉ+I2+H2O=SO42ˉ+2HI
2S2O32-+I2=S4O62-+2I-(S2O32-+1/2I2=1/2S4O62-+I-)
在250ml碘量瓶中加入10ml0.1NI2标准溶液和约10ml去离子水,称1g左右的干石膏,精确至0.0001g,加入其中。
滴加0.1mol/l的硫酸10ml,然后搅拌5分钟,此时混合物PH值在1-2之间,再加入100ml去离子水,用0.1NNa2S2O3标准溶液滴定至溶液为淡黄色,加入2ml淀粉溶液,再继续滴定至蓝色消失,期间用玻璃棒不断搅拌。
=0.6457*(V1-V2)/M
式中:
X—石膏中CaSO3·0.5H2O量,﹪
V1—I2标准溶液加入量,ml
V2—Na2S2O3标准溶液滴定消耗量,ml
M—石膏重量,g
2.4石膏中CaCO3含量的测定
方法原理:
石膏中CaCO3与过量盐酸反应,通过搅拌驱除溶液中生成的CO2,然后用用NaOH标准溶液反滴剩余的盐酸,即可算出石膏中总碳酸盐的含量。
样品中亚硫酸盐对试验的影响,通过加入双氧水氧化为硫酸盐而消除。
本试验涉及的化学反应:
CaCO3+2HCl=CaCl2+CO2↑+H2O
HCl+NaOH=NaCl+H2O
SO32-+H2O2=SO42-+H2O
称约1g的石膏,精确至0.0001g,放入300ml烧杯中,加入100ml去离子水和1ml30%的双氧水,2分钟后,加入20ml0.1NHCl标准溶液和20ml去离子水,在50-70℃放置大约15分钟。
加2滴酚酞指示剂,用0.1NNaOH标准溶液滴定至溶液由无色变为粉红色为终点,或用PH表测定,滴定PH为7,至终点。
X=(V1-V2)×0.1×100.09×100×0.001/(2×M)
=0.50045*(V1-V2)/M
式中:
X——石膏中CaCO3含量,%
V1——HCl标准溶液加入量,ml
V2—-NaOH标准溶液滴定消耗量,ml
M—-石膏量重量,g
2.5石膏中氯含量的测定
方法原理:
使固体石膏中的Cl-在水中充分溶出,然后用AgNO3滴定测得其含量。
称取10g石膏,精确至0.0001g,放入800ml烧杯,加入400ml热去离子水,加热搅拌10分钟,用定量滤纸过滤,沉淀用热去离子水洗涤,用容量瓶定容500ml。
吸取100ml滤液,加0.5ml30%的双氧水摇匀,2分钟后加1ml铬酸钾指示剂,用1ml/1mg滴定度AgNO3标准溶液滴定至溶液由黄色刚好出现砖红色为终点。
消耗AgNO3溶液G[ml]。
X=G×5×0.1/M
式中:
X—氯含量,﹪
G—AgNO3标准溶液消耗量,ml
V—滤液总体积500ml
V1—分析用滤液体积100ml
M—石膏重量,g
2.6石膏pH测定
室温下,在90ml去离子水中加入10g±0.1g的干石膏,搅拌该浓度为10%的溶液1分钟,随后静置5分钟。
开始测试前,在室温下用缓冲溶液校正PH表。
2.7.酸不溶物的测定
(1)试剂
A:
盐酸(1+5):
将1体积的盐酸与5体积的水混合
B:
1%的硝酸银溶液:
将1克硝酸银溶于90毫升水中,加入10毫升硝酸混匀
(2)试验步骤
准确称取试样约0.5克,置于250毫升烧杯中,用水润湿后盖上表面皿。
从杯口慢慢加入40毫升盐酸(1+5),待反应停止后,用水冲洗表面皿及杯壁并稀释至约75毫升。
加热煮沸3-4分钟,用定量滤纸过滤,以热水洗至无氯根为止。
将沉淀和滤纸一并移入已灼烧恒重的瓷坩埚中,在电炉上灰化,在950-1000℃的高温炉中灼烧20分钟,取出放入干燥器中冷却至室温称量
(3)结果计算
酸不溶物的百分含量(X2)按下式计算:
X2=100×G1/G
式中:
G1----灼烧后残渣重量,克
G----试样质量,克
三、吸收塔浆液的分析
(一)、吸收塔浆液的化学分析流程:
过滤浆液分离出固体物,烘干除去游离水,分析固体物中硫酸根、亚硫酸根、碳酸根;对滤液中测定氯根、亚硫酸根。
(二)、试验方法
3.1吸收塔浆液的pH测定:
在吸收塔就地取浆液,用便携式pH计测试其pH。
3.2吸收塔浆液密度的测试
取一准确体积带盖且有溢流口的不锈钢密度杯称取其重量B[g],将约100mL浆液加入密度杯中,采样迅速,要保持浆液大流量,冲洗密度杯外壁,擦拭干净,再称取其重量A[g]。
计算公式如下:
3.3吸收塔浆液的含固量的测定
真空过滤器称取其重量A(精确至0.1g),用该过滤器对V[mL]吸收塔浆液进行真空过滤,用碘量瓶收集滤液作浆液的液相项目试验。
滤饼在45℃下干燥一昼夜至恒重,称其重量B(精确至0.01g)。
计算公式如下:
浆液质量百分浓度=(B-A)/浆液1.2中(A-B)
3.4吸收塔浆液固体中CaCO3含量的测定
取1.3中的干燥固体,用研磨器研磨至粉状。
称约1g的滤饼,精确至0.0001g,放入300ml烧杯中,加入100ml去离子水和1ml30%的双氧水,2分钟后,加入20ml0.1NHCl标准溶液和20ml去离子水(CaCO3含量较高,加入40ml0.1NHCl标准溶液),在50-70℃放置大约15分钟。
加2滴酚酞指示剂,用0.1NNaOH标准溶液滴定至溶液由无色变为粉红色为终点,或用PH表测定,滴定PH为7,至终点。
X=(V1-V2)×0.1×100.09×100×0.001/(2×M)
=0.50045*(V1-V2)/M
式中:
X——滤饼中CaCO3含量,%
V1——HCl标准溶液加入量,ml
V2—-NaOH标准溶液滴定消耗量,ml
M—-滤饼量重量,g
3.5吸收塔浆液固体中CaSO4·2H2O含量的测定
3.5.1重量法
取3.3中的干燥固体,称取A克(约1g,精确至0.1mg)并将其放入250mL烧杯中,加入100mL去离子水和10mL浓盐酸,加热至沸腾并保持30min。
用中速定量滤纸过滤酸解液,并用大量热去离子水洗涤滤纸,直至用pH试纸检验过滤液为中性为止。
待完全冷却后将滤液定容至500mL,留待测试用。
试剂:
a)10wt%BaCl2溶液
b)浓盐酸
c)1wt%AgNO3溶液
测试方法:
在250mL烧杯中加入100mL酸解液,并加入50mL去离子水和5mL浓盐酸,将该溶液加热至沸腾。
然后逐滴滴入10mL10wt%BaCl2溶液,期间溶液保持沸腾。
将溶液静置4h以上(彻夜更好)。
用中速定量滤纸过滤溶液,并大量热去离子水冲洗沉淀物,直至用1wt%AgNO3溶液检验滤液无氯离子存在为止。
取一陶瓷坩锅在800℃下灼烧至恒重,称取重量B(精确至0.1mg)。
将滤纸和沉淀物移置该坩锅中,在800℃下灼烧至恒重,并称取其重量C(精确至0.1mg)。
计算公式如下:
3.5.2容量法
3.5.2.1、实验原理:
在石膏样品的溶解过程中加入氧化剂过氧化氢将其中的亚硫酸盐氧化为硫酸盐,同时加入阳离子交换树脂除去水中的阳离子。
制备好的溶液过滤后量取适量体积以氯化钡标准溶液进行滴定,(指示剂为偶氮胂Ⅲ)当溶液恰好由玫瑰红变成亮紫色时终点到达,根据氯化钡标准溶液消耗的体积,计算出样品中的硫酸盐含量(滴定后的硫酸盐含量还应扣除其中由亚硫酸盐氧化为硫酸盐的部分)。
反应方程如下:
CaSO3+H2O2=CaSO4+H2O
CaSO4+BaCL2=CaCL2+BaSO4
3.5.2.2、实验仪器:
万分之一电子天平、400ml玻璃烧杯、500ml容量瓶、真空过滤装置、滤纸、阳离子交换树脂、玻璃棒、50ml刻度量筒、50ml滴定管、磁力搅拌器,磁力搅拌棒。
3.5.2.3、试剂配制及标定:
3.5.2.3.1指示剂偶氮胂Ⅲ:
用烧杯称取0.5g偶氮胂Ⅲ,用少量除盐水溶解,移入1000ml容量瓶中,多次冲洗烧杯并移入容量瓶,加入除盐水稀释至刻度,摇匀。
3.5.2.3.2氯化钡溶液(0.005mol/L):
称取在110℃下干燥过2-4小时的BaCL21.0413g(BaCL2分子量208.26),放于干净的烧杯中,用100ml除盐水溶解,移入1000ml容量瓶中,再用除盐水清洗烧杯,并将清洗液移入1000ml容量瓶中,加除盐水至刻度。
混匀后存放于试剂瓶中待用。
3.5.2.3.3氯化钡溶液(0.005mol/L)的标定:
已知纯度的无水硫酸钠在120℃下烘干1小时。
精确称取硫酸钠0.1000—0.1200克2份,用少量无离子水溶解后,分别移人lO00ml容量瓶中,稀至刻度,摇匀。
把离子交换柱内的液面调至与树脂面平齐,注满硫酸钠溶液,液面高出“0”刻度约3mL,以每秒2—3滴的流速调节液面在“0”刻度处,弃去流出液。
交换柱下置50mL三角瓶以同样的流速准确取1.00-5.00mL(Vso-4)。
取样体积(Vso-4)的多少视SO-4,浓度而定,以此调节BaCl标准溶液消耗的体积不超过5.00mL。
对SO-4浓度大的基准溶液(或水样)应减少取样体积,但不小于1.00mL;对SO-4浓度小的应增加取样体积,但不超过10mL。
往三角瓶中加入25—30mL乙醇和4滴偶氮肿Ⅲ指示剂,用0.005mol/LBaC12标准
溶液边搅拌边滴定,溶液颜色由玫瑰红色变为紫色即为终点。
标定结果按下式计算:
MBa=WPVSO42-×1000/142.04VBaV
式中
MBa——BaCl2标准溶液的摩尔浓度,mol/L;
W—Na2SO4的质量,g;
P—Na2SO4的纯度,%;
VSO42---Na2SO4溶液的体积,mL;
VBa-BaC12消耗的体积,mL;
V一配制Na2SO4溶液的总体积,mL。
3.5.2.4、实验步骤:
3.5.2.4.1样品液制备:
准确称量0.2500g样品B倒入烧杯内,加入10ml30%H2O2和300ml除盐水;放入磁力搅拌棒搅拌10分钟;加入三勺阳离子交换树脂,再继续搅拌10分钟;将样品连同树脂倒入漏斗中过滤,滤入500ml容量瓶内,再用除盐水仔细冲洗数遍,稀释至刻度;
3.5.2.4.2样品测定:
取水样5.00mL,加入30ml乙醇和4滴偶氮胂Ⅲ,一边磁力搅拌一边用0.005mol/LBaC12进行滴定直至颜色由玫瑰红变成蓝紫色,记下消耗的氯化钡的体积A。
3.5.2.4.3计算:
SO42-(mmol/g)=0.005*A*100/B-SO32-
CaSO4·2H2O(%)=SO42-(mmol/g)*172.17*0.001*100
3.6吸收塔浆液固体中CaSO3·1/2H2O含量的测定
取1.3中用研磨器研磨至粉状的干燥滤饼样品
在250ml碘量瓶中加入10ml0.1NI2标准溶液和约10ml去离子水,称1g左右的干石膏,精确至0.0001g,加入其中。
滴加0.1mol/l的硫酸10ml,然后搅拌5分钟,此时混合物PH值在1-2之间,再加入100ml去离子水,用0.1NNa2S2O3标准溶液滴定至溶液为淡黄色,加入2ml淀粉溶液,再继续滴定至蓝色消失,期间用玻璃棒不断搅拌。
/1000
=0.6457*(V1-V2)/M
式中:
X—滤饼中CaSO3·0.5H2O量,﹪
V1—I2标准溶液加入量,ml
V2—Na2S2O3标准溶液滴定消耗量,ml
m—滤饼重量,g
3.7吸收塔浆液滤液中氯离子的测定
将1.3中的滤液取5ml加入250ml锥形瓶中,加去离子水100ml,加0.5ml30%的双氧水摇匀,2分钟后加1ml铬酸钾指示剂,用1ml/1mg滴定度AgNO3标准溶液滴定至溶液由黄色刚好出现砖红色为终点。
消耗AgNO3溶液B[mL]。
计算公式如下:
CL-(mg/l)=200*B
3.8吸收塔浆液滤液中亚硫酸根的测定
3.8.1试剂:
a)0.1N的H2SO4
b)0.1N的I2标准溶液
c)0.1N的Na2S2O3标准溶液
3.8.2测试方法:
将10mL0.1NI2溶液和10mL去离子水加入250mL烧杯中,滴加0.1mol/l的硫酸10ml,取20mL吸收塔浆液滤液加入烧杯中,用磁力搅拌器搅拌5min。
,此时混合物PH值在1-2之间,再加入100ml去离子水,用0.1NNa2S2O3标准溶液滴定至溶液为淡黄色,加入2ml淀粉溶液,再继续滴定至蓝色消失,期间用玻璃棒不断搅拌,记下Na2S2O3的消耗体积V[mL]。
计算公式如下:
3.9吸收塔浆液滤液中氟离子浓度(离子选择电极法GB/T15555.11-1995)
3.9.1实验原理
氟离子选择电极是将氟化镧单晶(掺入微量氟化铕(Ⅱ)以增加导电性)封在塑料管的一端,管内装0.1mg/LNaF和0.1mg/LNaCl溶液,以Ag-AgCl电极为参比电极,构成氟离子选择电极。
用氟离子选择电极测定水样时,以氟离子选择电极作指示电极,以饱和甘汞电极作参比电极,组成的电池为:
氟离子选择电极∣试液∣∣SCE
如果忽略液接电位,电池的电动势为:
E=b-0.0592㏒a
即电池的电动势与试液中氟离子活度的对数成正比。
氟离子选择电极一般在1~10-6mg/L范围内符合能斯特方程式。
氟离子选择电极具有较好的选择性。
常见阴离子NO3-,SO42-、PO43-、Ac-、Cl-、Br-、I-、HCO3-等不干扰,主要干扰物是OH-。
产生干扰的原因,很可能是由于在膜表面发生如下反应:
LaF3+3OH-→La(OH)3+3F-
反应产物F-因电极本身的响应而造成干扰。
在较高酸度时由于形成HF2-而降低F-的离子活度,因此测定时,需控制试液的PH在5~6之间通常用乙酸缓冲溶液控制.常见阳离子除易与F-形成稳定配位离子的Fe3+、Al3+、Sn(Ⅳ)干扰外其他不干扰。
这几种离子的干扰可加入柠檬酸钠进行掩蔽。
用氟离子选择电极测定的是溶液中离子的活度,因此必须加入大量电解质控制溶液的离子强度。
氟离子选择电极测定氟离子时,应加入总离子强度调节缓冲液,以控制溶液pH和离子强度以消除干扰。
3.9.2仪器与试剂
离子计;氟离子选择电极;饱和甘汞电极;电磁搅拌器;100mL容量瓶七只;100mL烧杯两个;10mL移液管。
0.1000mol/LF标准溶液;总离子强度调节缓冲液(TISAB)。
3.9.3实验步骤
3.9.3.1.氟离子选择电极的准备
氟离子选择电极在使用前,应放在含10-4mol/L的F或更低浓度的F-溶液中浸泡,活化约30分钟。
使用时,先用去离子水吹洗电极,再在去离子水中洗至电极的纯水电位。
其方法是将电极浸入去离子水中,在离子计上测量其电位,然后更换去离子水,观察其电位变化,如此反复进行处理,直至其电位达到稳定并为它的纯水电位为止。
3.9.3.2.测量范围及能斯特斜率的测量:
在5只100mL容量瓶中用10mL移液管移取0.1000mol/LF-标准溶液与第一只100mL容量瓶中,加入TISAB10mL,去离子水稀释至标线,摇匀,配成1.00×10-2mol/LF标准溶液;在第二只100mL容量瓶中,加入1.00×10-2mol/LF-标准溶液10mL和TISAB10mL,去离子水稀释至标线,摇匀,配成1.00×10-3mol/LF-标准溶液。
按上述方法依次配制1.00×10-4--1.00×10-6mol/LF-标准溶液。
将适量F-标准溶液(浸没电极即可)分别倒入5只塑料烧杯中,放入磁性搅拌子,插入氟离子选择电极和饱和甘汞电极,连接好离子计,开启电源搅拌器,依稀至浓依次测量,在仪器数字显示在±1mV内,读取电位值,记录数据。
再分别测定其他F-标准溶液的电位值。
3.9.3.3.氟含量的测定:
⑴.标准曲线法:
在实验室接取50.0mL水样置于100mL容量瓶中,加入TISAB10mL,去离子水稀释至标线,摇匀。
全部到入一烘干的烧杯中,按2中方法测定其电位值,并记录数据。
平行测三份。
⑵标准加入法:
在⑴测量后加入1.00mL1.00×10-3mol/LF-标准溶液,再测定其电位值并记录数据。
3.9.4.实验数据记录及处理:
㈠标准曲线法:
1.标准溶液的电势记录:
(25℃)
电势/mV
浓度对数
-328.0
-6(1×10-6mol/l)
-299.2
-5(1×10-5mol/l)
-246.0
-4(1×10-4mol/l)
-187.8
-3(1×10-3mol/l)
-130.0
-2(1×10-2mol/l)
浓度对数与电势的关系曲线:
2.实验测得水样的电势为:
(以自来水为例)
实验编号
1
2
3
所测电势/mV
-294.6
-305.0
-307.9
求得电势平均值为:
Ex=-302.5mV
测量的标准偏差:
=0.146
相对标准偏差:
=0.28%
由曲线可知,电势与浓度的关系为:
E=50.74㏒C-35.24
3.自来水中F-浓度的计算:
将Ex=-302.5mV带入上式,得经过处理的溶液中F-浓度为:
Cx=1.0×10-5.27mol/L=2.93×10-6mol/L.
所以,自来水中F-浓度为:
Cx=5.86×10-6mol/L.
㈡.标准加入法:
1.所测得的加入了F-标准溶液的溶液的电势:
实验编号
1
2
3
所测电势/mV
-291.9
-292.8
-291.0
2.求得平均值为:
-291.9mV
测量的标准偏差:
=0.09
相对标准偏差:
=0.005%
△E=-291.9mV+302.5mV=10.60mV
所以,Cx=△C/(10△E/S-1)△C=Vs*Cs/100
式中,Vs、Cs分别为加入的F-标准液的体积(mL)和浓度(mol/L),S为实验所测得的电极的能斯特响应斜率。
代入数据可得Cx=2.88×10-6mol/L.
所以,自来水中F-的浓度为:
Cx=5.76×10-6mol/L
3.误差分析:
实验中由于充分考虑到了各种可能造成误差的因素,因此,结果所产生的误差较小。
但由于仪器的问题,使得所测的电位有较大误差,不过因最终要的是线性关系曲线,故对实验结果造成的影响可不计。
3.9.5注意事项:
1.测量标准溶液时,浓度应由稀至浓,每次测定后用被测试液清洗电极、烧杯及搅拌子。
2.绘制标准曲线时,测定一系列标准溶液后,应将电极清洗至原空白电位值,然后再测定未知液的电位。
3.测定过程中,更换溶液时,测量键应断开,以免损坏离子计。
4.测定过程中,搅拌溶液的速度应恒定。
5.氟离子电极暂不用时,宜于干放。
6.用氟离子选择电极法测定自来水中氟离子含量时,加入TISAB的组成和作用:
TISAB的组成成分是0.1mol/L氯化钠溶液,0.25mol/LHAc-0.75mol/LNaAc溶液和0.001mol/L柠檬酸钠溶液,其作用分别是控制离子强度,控制溶液的酸度,使pH=5-6,和掩蔽自来水中含有的Al3+、Fe2+、Sn4+等干扰离子,防止F-与金属离子形成配合物
7.标准曲线法和标准加入法两种方法比较:
(1).标准曲线法:
可以适用于多次测量,并且要求标准溶液和样品具有恒定的离子强度,并维持在适宜的pH范围内.调节离子强度所用电解质不应对测定有干扰,调节离子强度的溶液,也常加入适当的络合剂或其他试剂以消除干扰离子的影响。
(2).标准加入法:
是在其他组分共存情况下进行测量的,因此实际上减免了共存组分的影响,古这种方法适合于成分不明或是组成复杂的试样的测定。
8、最小二乘法确定斜率
3.10氟的测定(分光光度法)
3.10.1方法提要
试样经碱熔后用水浸取,在锌盐存在下,将pH调至8-9以分离干扰离子,经干过滤后,分取部分滤液于pH4.5的34%丙酮介质中形成镧-茜素络合剂-氟三元络合物,进行比色。
3.10.2试剂与仪器
a.碳酸钠(固体)
b.氢氧化钠(固体)。
c.硝酸锌溶液:
50克氧化锌溶于300毫升硝酸(1+1),以水稀释至1升。
d.乙酸-乙酸钠缓冲溶液(pH4.0-4.2):
称取60克乙酸钠(NaAC·3H2O)溶于水中,加入115毫升冰乙酸,以水稀释至1升,摇匀(用pH计或精密试纸检验)。
e.0.04M氯化镧溶液:
称取1.6292克氧化镧(La2O3,分析纯),加少量水,在搅拌下滴加盐酸(1+1)加热溶解,冷却后用水稀释至250毫升。
f.显色剂:
称取0.0965克茜素络合剂(简称ALC),用数滴水湿润,加入氨水(1+1)1毫升缓慢溶解,依次加丙酮125毫升,pH4.0-4.2的缓冲溶液50毫升,0.04M氯化镧溶液6.5毫升,用水稀释至250毫升,摇匀(此溶液至少能稳定一周)。
g.1%酚酞指示剂乙醇溶液:
将1克酚酞溶于100毫升乙醇中。
h.2%氢氧化钠溶液:
2克氢氧化钠溶于100毫升水中保存于塑料瓶内。
i.乙酸(1+8):
将1体积的乙酸与8体积水混合。
j.氟标准溶液(A):
准确称取0.2210克氟化钠(NaF,优级纯,置于铂坩埚内已于120℃烘过2小时)于300毫升烧杯中,用水溶解后移入1000毫升容量瓶中,用水稀释至标线,摇匀。
贮存于塑料瓶内,此溶液每毫升含有0.1
 升级会员
升级会员 