西北工业大学《材料科学基础》课后题答案.doc
《西北工业大学《材料科学基础》课后题答案.doc》由会员分享,可在线阅读,更多相关《西北工业大学《材料科学基础》课后题答案.doc(36页珍藏版)》请在冰豆网上搜索。
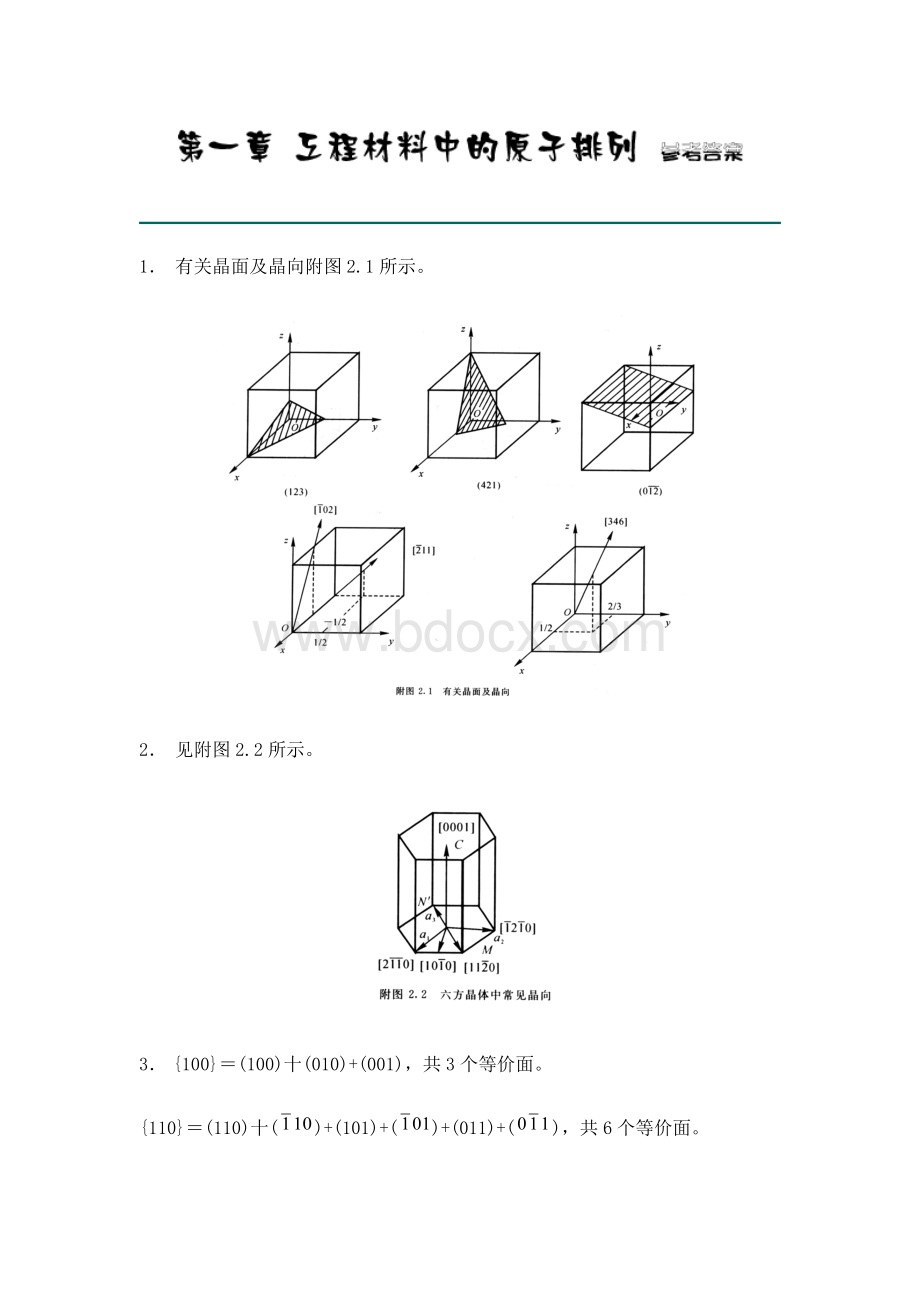
1. 有关晶面及晶向附图2.1所示。
2. 见附图2.2所示。
3. {100}=(100)十(010)+(001),共3个等价面。
{110}=(110)十()+(101)+()+(011)+(),共6个等价面。
{111}=(111)+()+()+(),共4个等价面。
共12个等价面。
4. 单位晶胞的体积为VCu=0.14nm3(或1.4×10-28m3)
5.
(1)0.088nm;
(2)0.100nm。
6. Cu原子的线密度为2.77×106个原子/mm。
Fe原子的线密度为3.50×106个原子/mm。
7. 1.6l×l013个原子/mm2;1.14X1013个原子/mm2;1.86×1013个原子/mm2。
8.
(1)5.29×1028个矽原子/m3;
(2)0.33。
9. 9.0.4×10-18/个原子。
10. 1.06×1014倍。
11.
(1)这种看法不正确。
在位错环运动移出晶体后,滑移面上、下两部分晶体相对移动的距离是由其柏氏矢量决定的。
位错环的柏氏矢量为b,故其相对滑移了一个b的距离。
(2)A'B'为右螺型位错,C'D'为左螺型位错;B'C'为正刃型位错,D'A'为负刃型位错。
位错运动移出晶体后滑移方向及滑移量如附图2.3所示。
12.
(1)应沿滑移面上、下两部分晶体施加一切应力τ0,的方向应与de位错线平行。
(2)在上述切应力作用下,位错线de将向左(或右)移动,即沿着与位错线de垂直的方向(且在滑移面上)移动。
在位错线沿滑移面旋转360°后,在晶体表面沿柏氏矢量方向产生宽度为一个b的台阶。
13.
(1),其大小为,其方向见附图2.4所示。
(2)位错线方向及指数如附图2.4所示。
14.
(1)能。
几何条件:
∑b前=∑b后=;能量条件:
∑b前2=>∑b后2=
(2)不能。
能量条件:
∑b前2=∑b后2,两边能量相等。
(3)不能。
几何条件:
∑b前=a/b[557],∑b后=a/b[11¯1],不能满足。
(4)不能。
能量条件:
∑b前2=a2 <∑b后2=,即反应后能量升高。
15.
(1)能够进行。
因为既满足几何条件:
∑b前=∑b后=,又满足能量条件:
∑b前2=>∑b后2=
(2)b合=;该位错为弗兰克不全位错。
16.
(1)假设晶体中位错线互相缠结、互相钉扎,则可能存在的位错源数目个/Cm3。
(2)τNi=1.95×107Pa。
17. 当θ=1°,D=14nm;θ=10°,D=1.4nm时,即位错之间仅有5~6个原子间距,此时位错密度太大,说明当θ角较大时,该模型已不适用。
18. 畸变能是原来的0.75倍(说明形成亚晶界后,位错能量降低)。
19. 设小角度晶界的结构由刃型位错排列而成,位错间距为D。
晶界的能量γ由位错的能量E构成,设l为位错线的长度,由附图2.5可知,
由位错的能量计算可知,
取R=D(超过D的地方,应力场相互抵消),r0=b和θ=b/D代入上式可得:
式中
20.
(1)晶体点阵也称晶体结构,是指原子的具体排列;而空间点阵则是忽略了原子的体积,而把它们抽象为纯几何点。
(2)密排六方结构。
(3)原子半径发生收缩。
这是因为原子要尽量保持自己所占的体积不变或少变[原子所占体积VA=原子的体积(4/3πr3+间隙体积],当晶体结构的配位数减小时,即发生间隙体积的增加,若要维持上述方程的平衡,则原子半径必然发生收缩。
(4)不能。
因为位错环是通过环内晶体发生滑移、环外晶体不滑移才能形成。
(5)外力在滑移面的滑移方向上的分切应力。
(6)始终是柏氏矢量方向。
(7)位错的交割。
(8)共格界面。
(9)否,扭转晶界就由交叉的同号螺型位错构成。
1. 其比较如附表2.1所示。
附表2.1 间隙固溶体与间隙化合物的比较
类 别
间隙固熔体
间隙化合物
相同点
一般都是由过渡族金属与原子半径较小的C,N,H,O,B等非金属元素所组成
不同点
晶体结构
属于固熔体相,保持熔剂的晶格类型
属于金属化合物相,形成不同于其组元的新点阵
表达式
用α、β、γ等表示
用化学分子式MX,M2X等表示
机械性能
强度、硬度较低,塑性、韧性好
高硬度、高熔点,甥性、韧性差
2. 有序固熔体,其中各组元原子分别占据各自的布拉菲点阵——称为分点阵,整个固熔体就是由各组元的分点阵组成的复杂点阵,也叫超点阵或超结构。
这种排列和原子之间的结合能(键)有关。
结合能愈大,原子愈不容易结合。
如果异类原子间结合能小于同类原子间结合能,即EAB<(EAA十EBB)/2,则熔质原子呈部分有序或完全有序排列。
有序化的推动力是混合能参量(εm=εAB-1/2(EAA+EBB))εm<0,而有序化的阻力则是组态熵;升温使后者对于自由能的贡献(-TS)增加,达到某个临界温度以后,则紊乱无序的固熔体更为稳定,有序固熔体消失,而变成无序固熔体。
3. 在原子尺寸因素相近的情况下,上述元素在Ag中的固熔度(摩尔分数)受原子价因素的影响,即价电子浓度e/a是决定固熔度(摩尔分数)的一个重要因素。
它们的原子价分别为2,3,4,5价,Ag为1价,相应的极限固熔度时的电子浓度可用公式c=ZA(1一xB)+ZBxB
计算。
式中,ZA,ZB分别为A,B组元的价电子数;xB为B组元的摩尔分数。
上述元素在固溶度(摩尔分数)极限时的电子浓度分别为1.43,1.42,1.39,1.31。
4. Α-Fe为体心立方点阵,致密度虽然较小,但是它的间隙数目多且分散,因而间隙半径很小:
r四=0.291,R=0.0361nm;r八=0.154,R=0.0191nm。
H,N,C,B等元素熔人。
α-Fe中形成间隙固熔体,由于尺寸因素相差很大,所以固熔度(摩尔分数)都很小。
例如N在α-Fe中的固熔度(摩尔分数)在590℃时达到最大值,约为WN=0.1/l0-2,在室温时降至WN=0.001/l0-2;C在α-Fe中的固溶度(摩尔分数)在727℃时达最大值,仅为WC=0.02l8/10-2,在室温时降至WC=0.006/10-2。
所以,可以认为碳原子在室温几乎不熔于α-Fe中,微量碳原子仅偏聚在位错等晶体缺陷附近。
假若碳原子熔入。
α-Fe中时,它的位置多在α-Fe的八面体间隙中心,因为。
α-Fe中的八面体间隙是不对称的,形为扁八面体,[100]方向上间隙半径r=0.154R,而在[110]方向上,r=0.633R,当碳原子熔入时只引起一个方向上的点阵畸变。
硼原子较大,熔人间隙更为困难,有时部分硼原子以置换方式熔人。
氢在α-Fe中的固熔度(摩尔分数)也很小,且随温度下降时迅速降低。
以上元素在γ-Fe。
中的固熔度(摩尔分数)较大一些。
这是因为γ-Fe具有面心立方点阵,原子堆积致密,间隙数目少,故间隙半径较大:
rA=0.414,R=0.0522nm;r四=0.225,R=0.0284nm。
故上述原子熔入时均处在八面体间隙的中心。
如碳在γ-Fe中最大固熔度(质量分数)为WC=2.1l/10-2;氮在γ-Fe中的最大固熔度(质量分数)约为WN=2.8/10-2。
5. 密度ρ=5.97g/cm3。
6. 两离子的中心距离为0.234nm。
7. 碳原子占据10.2%的八面体间隙位置;氮原子占据12.5%的八面体间隙位置。
8. 这是因为形成固熔体时,熔质原子的熔入会使熔剂结构产生点阵畸变,从而使体系能量升高。
熔质与熔剂原子尺寸相差越大,点阵畸变的程度也越大,则畸变能越高,结构的稳定性越低,熔解度越小。
一般来说,间隙固熔体中熔质原子引起的点阵畸变较大,故不能无限互溶,只能有限熔解。
9. 9
(1)0.278nm;
(2)0.393nm(3)0.482nm;(4)0.622nm;(5)0.393nm。
10.
(1)WLi+=16/10-2,WMg2+=24/1020,WF-=44/10-2,WO2—=16/10-2
(2)该固熔体的密度ρ=2.9g/cm3。
11. 故理论强度介于之间,即4900~7000MPa
12. 模子的尺寸l=15.0mm。
13.
故可能是丙酮。
14. 画出丁醇(C4H9OH)的4种可能的异构体如下:
15.
(1)单体质量为12X2+1X2+35.5X2=97g/mol;
(2)聚合度为 n=60000/97=620。
16.
(1)均方根据长度4.65nm;
(2)分子质量m=7125g。
17. 理论上的最大应变为3380%。
18. 单体的摩尔分数为:
X苯烯=20/10-2,X丁二烯=40/10-2,X丙烯晴=40/10-2
19.
(1)和
(2)如下:
(3)每摩尔的水(0.6X1024)形成时,需要消去0.6X1024的C—O及N—H键,同时形成0.6X1024的C—N及H—O键。
净能量变化为-15kJ/mol。
20. 硅酸盐结构的基本特点:
(1)硅酸盐的基本结构单元是[Si04]四面体,硅原子位于氧原子四面体的间隙中。
硅—氧之间的结合键不仅是纯离子键,还有相当的共价键成分。
(2)每一个氧最多只能被两个[Si04]四面体所共有。
(3)[Si04]四面体可以是互相孤立地在结构中存在,也可以通过共顶点互相连接。
(4)Si—O--Si的结合键形成一折线。
硅酸盐分成下列几类:
(1)含有有限硅氧团的硅酸盐;
(2)链状硅酸盐;
(3)层状硅酸盐;
(4)骨架状硅酸盐。
21. 因为大多数陶瓷主要由晶相和玻璃相组成,这两种相的热膨胀系数相差较大,由高温很快冷却时,每种相的收缩不同,所造成的内应力足以使陶瓷器件开裂或破碎。
22. 陶瓷材料中主要的结合键是离子键及共价键。
由于离子键及共价键很强,故陶瓷的抗压强度很高,硬度极高。
因为原子以离子键和共价键结合时,外层电子处于稳定的结构状态,不能自由运动,故陶瓷材料的熔点很高,抗氧化性好,耐高温,化学稳定性高。
1. 分析结晶相变时系统自由能的变化可知,结晶的热力学条件为∆G<0;由单位体积自由能的变化可知,只有∆T>0,才有∆GB<0。
即只有过冷,才能使∆G<0。
动力学条件为液—固界面前沿液体的温度T由临界晶核形成功A=1/3σS可知,当形成一个临界晶核时,还有1/3的表面能必须由液体中的能量起伏来提供。
液体中存在的结构起伏,是结晶时产生晶核的基础。
因此,结构起伏是结晶过程必须具备的结构条件。
2. 凝固驱动力∆G=一3253.5J/mol。
3.
(1)rk=9.03X10-10m;
(2)n=261个。
4. 所谓界面的平衡结构,是指在界面能最小的条件下,界面处于最稳定状态。
其问题实质是分析当界面粗糙化时,界面自由能的相对变化。
为此,作如下假定:
(1) 液、固相的平衡处于恒温条件下;
(2) 液、固相在界
 升级会员
升级会员 