Realtime PCR.docx
《Realtime PCR.docx》由会员分享,可在线阅读,更多相关《Realtime PCR.docx(18页珍藏版)》请在冰豆网上搜索。
RealtimePCR
实时荧光定量PCR:
一种科学准确的定量方法
实时荧光定量PCR技术于1996年由美国AppliedBiosystems公司推出,由于该技术不仅实现了PCR从定性到定量的飞跃,而且与常规PCR相比,它具有特异性更强、有效解决PCR污染问题、自动化程度高等特点,目前已得到广泛应用。
本文试就其定量原理、方法及参照问题作一介绍。
一.实时荧光定量PCR原理
所谓实时荧光定量PCR技术,是指在PCR反应体系中加入荧光基团,利用荧光信号积累实时监测整个PCR进程,最后通过标准曲线对未知模板进行定量分析的方法。
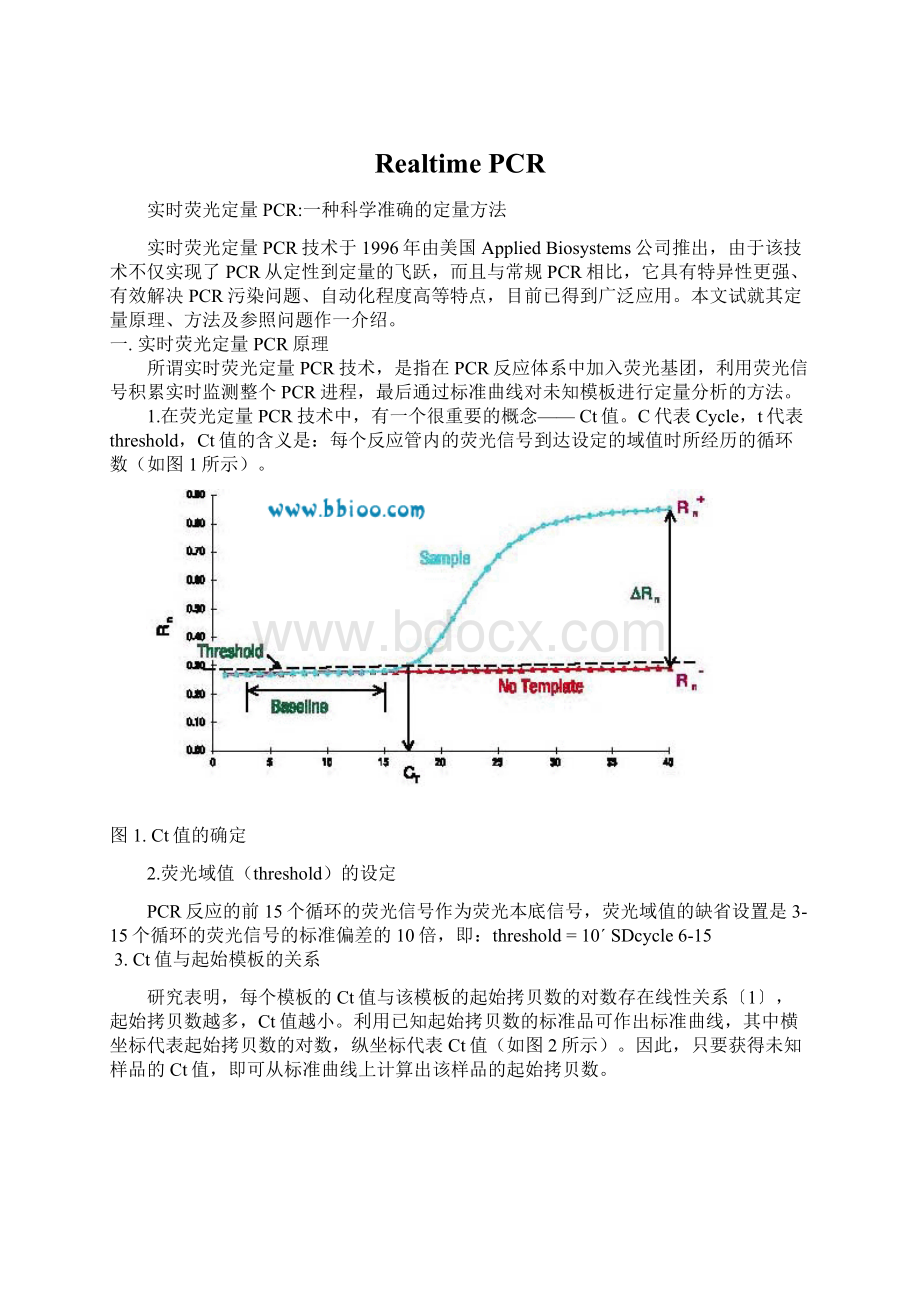
1.在荧光定量PCR技术中,有一个很重要的概念——Ct值。
C代表Cycle,t代表threshold,Ct值的含义是:
每个反应管内的荧光信号到达设定的域值时所经历的循环数(如图1所示)。
图1.Ct值的确定
2.荧光域值(threshold)的设定
PCR反应的前15个循环的荧光信号作为荧光本底信号,荧光域值的缺省设置是3-15个循环的荧光信号的标准偏差的10倍,即:
threshold=10´SDcycle6-15
3.Ct值与起始模板的关系
研究表明,每个模板的Ct值与该模板的起始拷贝数的对数存在线性关系〔1〕,起始拷贝数越多,Ct值越小。
利用已知起始拷贝数的标准品可作出标准曲线,其中横坐标代表起始拷贝数的对数,纵坐标代表Ct值(如图2所示)。
因此,只要获得未知样品的Ct值,即可从标准曲线上计算出该样品的起始拷贝数。
图2.荧光定量标准曲线
4.荧光化学
荧光定量PCR所使用的荧光化学可分为两种:
荧光探针和荧光染料〔2〕。
现将其原理简述如下:
1)TaqMan荧光探针:
PCR扩增时在加入一对引物的同时加入一个特异性的荧光探针,该探针为一寡核苷酸,两端分别标记一个报告荧光基团和一个淬灭荧光基团。
探针完整时,报告基团发射的荧光信号被淬灭基团吸收;PCR扩增时,Taq酶的5’-3’外切酶活性将探针酶切降解,使报告荧光基团和淬灭荧光基团分离,从而荧光监测系统可接收到荧光信号,即每扩增一条DNA链,就有一个荧光分子形成,实现了荧光信号的累积与PCR产物形成完全同步。
如图3所示。
图3.TaqMan荧光探针工作原理
2)SYBR荧光染料:
在PCR反应体系中,加入过量SYBR荧光染料,SYBR荧光染料特异性地掺入DNA双链后,发射荧光信号,而不掺入链中的SYBR染料分子不会发射任何荧光信号,从而保证荧光信号的增加与PCR产物的增加完全同步。
二.内标在传统定量中的意义
1.几种传统定量PCR方法简介〔3〕:
1)内参照法:
在不同的PCR反应管中加入已定量的内标和引物,内标用基因工程方法合成。
上游引物用荧光标记,下游引物不标记。
在模板扩增的同时,内标也被扩增。
在PCR产物中,由于内标与靶模板的长度不同,二者的扩增产物可用电泳或高效液相分离开来,分别测定其荧光强度,以内标为对照定量待检测模板〔4〕。
2)竞争法:
选择由突变克隆产生的含有一个新内切位点的外源竞争性模板。
在同一反应管中,待测样品与竞争模板用同一对引物同时扩增(其中一个引物为荧光标记)。
扩增后用内切酶消化PCR产物,竞争性模板的产物被酶解为两个片段,而待测模板不被酶切,可通过电泳或高效液相将两种产物分开,分别测定荧光强度,根据已知模板推测未知模板的起始拷贝数〔5〕。
3)PCR-ELISA法:
利用地高辛或生物素等标记引物,扩增产物被固相板上特异的探针所结合,再加入抗地高辛或生物素酶标抗体-辣根过氧化物酶结合物,最终酶使底物显色。
常规的PCR-ELISA法只是定性实验,若加入内标,作出标准曲线,也可实现定量检测目的〔6〕。
2.内标在传统定量中的作用
由于传统定量方法都是终点检测,即PCR到达平台期后进行检测,而PCR经过对数期扩增到达平台期时,检测重现性极差。
同一个模板在96孔PCR仪上做96次重复实验,所得结果有很大差异(见图4),因此无法直接从终点产物量推算出起始模板量。
加入内标后,可部分消除终产物定量所造成的不准确性。
但即使如此,传统的定量方法也都只能算作半定量、粗略定量的方法。
图4.相同模板在同一台PCR仪上进行96次扩增的扩增曲线图终点处检测产物量不恒定;Ct值则极具重现性
三.内标对荧光定量PCR的影响
1.实时荧光定量PCR无需内标实时荧光定量PCR技术有效地解决了传统定量只能终点检测的局限,实现了每一轮循环均检测一次荧光信号的强度,并记录在电脑软件之中,通过对每个样品Ct值的计算,根据标准曲线获得定量结果。
因此,实时荧光定量PCR无需内标是建立在两个基础之上的:
1)Ct值的重现性PCR循环在到达Ct值所在的循环数时,刚刚进入真正的指数扩增期(对数期),此时微小误差尚未放大,因此Ct值的重现性极好,即同一模板不同时间扩增或同一时间不同管内扩增,得到的Ct值是恒定的。
(见图4)
2)Ct值与起始模板的线性关系由于Ct值与起始模板的对数存在线性关系,可利用标准曲线对未知样品进行定量测定,因此,实时荧光定量PCR是一种采用外标准曲线定量的方法。
2.内标对实时荧光定量PCR的影响若在待测样品中加入已知起始拷贝数的内标,则PCR反应变为双重PCR,双重PCR反应中存在两种模板之间的干扰和竞争,尤其当两种模板的起始拷贝数相差比较大时,这种竞争会表现得更为显著〔7,8〕。
但由于待测样品的起始拷贝数是未知的,所以无法加入合适数量的已知模板作为内标。
也正是这个原因,传统定量方法虽然加入内标,但仍然只是一种半定量的方法。
美国Texas大学的科研人员进行了外标法定量和内标法定量的方法学比较,得出的结论是:
内标法作为定量或半定量的手段是不可靠的,而外标准曲线的定量方法是一种准确的、值得信赖的科学方法〔9〕。
AppliedBiosystems公司的7700型实时荧光定量PCR仪是全球公认的荧光定量PCR的金标准,可实现多色荧光同时检测,但定量检测仍然采用外标准曲线的方法,而不用内标进行定量。
综上所述,利用外标准曲线的实时荧光定量PCR是迄今为止定量最准确,重现性最好的定量方法,已得到全世界的公认,广泛用于基因表达研究、转基因研究,药物疗效考核、病原体检测等诸多领域〔10,11,12,13〕。
参考文献
1.HiguchiR,FocklerC,etal.KineticPCRanalysis:
real-timemonitoringofDNAamplificationreactions.Biotechnology,1993,11(9):
1026-1030.
2.DNA/RNAReal-TimeQuantitativePCR-Rev.BAppliedBiosystems
3.定量聚合酶链反应的研究进展与临床应用.中华检验医学杂志2000,23
(2):
120-121.
4.AndersonKM,CheungPH,KellMD.RapidgenerationofhomologousinternalstandardsandevaluationofdataforquantitaionofmessengerRNAbycompetitivepolymerasechainreaction.JPharmacolToxicolMethods,1997,38:
133-140.
5.WuhuA,WaiztuM,KochA.Arapidandsensitiveprotocolforcompetitivereversetranscriprase(CRT)PCRanalysisofcellulargenes.BrainPathol,1998,8:
13-18
6.仝文斌,高巍,费然等.核酸扩增产物的量化酶免疫通用型检测方法.中华医学检验杂志,1999,22:
83-86.
7.ActorJK,LimorJR,HunterRL.Aflexiblebioluminescent-quantitivepolymerasereactionassayforanalysisofcompetitivePCRamplicons.JClinLabAnal,1999,13
(1):
40-47.
8.SchnellS,MendozaC.Enzymologicalconsiderationsforatheoreticaldescriptionofthequantitativecompetitivepolymerasechainreaction(QC-PCR).JTheorBiol,1997,84(4):
433-440.
9.KeLD,ChenZ,YungWK.Areliabilitytestofstandard-basedquantitativePCR:
exogenousvsendogenousstandards.Mol.CellProbes,2000,14
(2):
127-135.
10.BeckerK.,D.PanandC.B.Whitely.Real-timequantitativepolymerasechainreactiontoassessgenetransfer.Hum.GeneTher,1999,10:
2559-2566.
11.Higgis,J.A.,etal.5’nucleasePCRassaytodetectYersiniapesits.JClinMicrobiol,1998,36:
2284-2288.
12.BiecheI.,P.Oondy,etal.Real-timereversetranscription-PCRassayforfuturemanagementofERBB2-basedclinicalapolications.Clin.Chem,1999,45:
1148-1156.
13.WangX.X.Li,etal.Applicationofreal-timepolymerasechainreactiontoquantitateinducedexpressionofinterleukin-1betamRNAinischemicbraintolerance.2000,J.Neurosci.Res,59
(2):
238-46.
定量PCRTaqman探针设计要领
作者:
未知来源:
时间:
2007-8-1
自90年代Taqman探针诞生以来,虽然荧光探针(引物)不断有新的技术出现,但是作为一种经典的定量PCR技术,Taqman探针技术仍然是许多实验研究人员进行定量检测的首选,这主要是因为相对于SYBR荧光染料,Taqman探针具有序列特异性,只结合到互补区,而且荧光信号与扩增的拷贝数具有一一对应的关系,因此特异性强灵敏度高,而且条件优化容易;而相对于杂交探针,Taqman探针只要设计一条探针,因此探针设计较便宜方便,而且也能完成基本的定量PCR要求。
当然Taqman定量方法由于还是要合成探针,也给实验操作带来了挑战。
一般Taqman定量PCR实验过程为:
目的基因查找比对→探针与引物设计→探针与引物合成→配置反应体系→反应参数→重复实验,优化条件→获得曲线数据,比对标准曲线→再重复验证。
第一步:
在第一步目的基因查找比对过程中可以利用NCBIgenbank序列以及DNAstar等软件完成目的DNA或者RNA的查找与比对——这在分析测序报告的时候相信很多人操作过,这一步需要注意的就是要保证所分析的序列在一个contig(重叠群,即染色体的一些区域中毗邻DNA片段重叠的情况)内。
第二步:
如果其它条件一致,那么这个第二步——引物探针的设计就可以说是定量PCR成败的关键了,通过各方面经验的总结有以下几个基本的原则:
总体原则
*先选择好探针,然后设计引物使其尽可能的靠近探针。
*所选序列应该高度特异,尽量选择具有最小二级结构的扩增片段——这是因为二级结构会影响反应效率,而且还会阻碍酶的扩增。
建议先进行二级结构检测,如果不能避免二级结构,那么就要相应提高退火温度。
*扩增长度应不超过400bp,理想的最好能在100-150bp内,扩增片段越短,有效的扩增反应就越容易获得。
较短的扩增片段也容易保证分析的一致性。
*保持GC含量在20%和80%之间,GC富含区容易产生非特异反应,从而会导致扩增效率的降低,以及出现在荧光染料分析中非特异信号。
*为了保证效率和重复性,应避免重复的核苷酸序列,尤其是G(不能有4个连续的G)
*将引物和探针互相进行配对检测,以避免二聚体和发卡结构的形成。
引物设计原则
*序列选取应在基因的保守区段
*避免引物自身或与引物之间形成4个或4个以上连续配对,避免引物自身形成环状发卡结构
*典型的引物18到24个核苷长。
引物需要足够长,保证序列独特性,并降低序列存在于非目的序列位点的可能性。
但是长度大于24核苷的引物并不意味着更高的特异性。
较长的序列可能会与错误配对序列杂交,降低了特异性,而且比短序列杂交慢,从而降低了产量。
*Tm值在55-65℃(因为60℃核酸外切酶活性最高),GC含量在40%-60%
*引物之间的TM相差避免超过2℃
*引物的3’端避免使用碱基A,引物的3’端避免出现3个或3个以上连续相同的碱基
*为避免基因组的扩增,引物设计最好能跨两个外显子。
*Taqman探针技术要求片段长度在50bp-150bp
*引物末端(最后5个核苷酸)不能有超过2个的G和C。
探针设计原则
*探针位置尽可能地靠近上游引物
*探针长度应在15-45bp(最好是20-30bp),以保证结合特异性
*检测探针的DNA折叠和二级结构
*Tm值在65-70℃,通常比引物TM值高5-10℃(至少要5℃),GC含量在40%-70%
*探针的5’端应避免使用G鸟嘌呤——因为5'G会有淬灭作用,而且即使是被切割下来还会存在淬灭作用。
*整条探针中,碱基C的含量要明显高于G的含量——G含量高会降低反应效率,这时就应选择配对的另一条链作为探针。
*为确保引物探针的特异性,最好将设计好的序列在blast中核实一次,如果发现有非特异性互补区,建议重新设计引物探针。
TaqmanMGB探针设计
*探针的5’端避免出现G,即使探针水解为单个碱基,与报告基团相相连的G碱基仍可淬灭基团的荧光信号。
*Tm值应为65-67℃。
*尽量缩短TaqmanMGB探针,但探针长度不少于13bp。
*尽量避免出现重复的碱基,尤其是G碱基,应避免出现4个或4个以上的G重复出现。
*原则上MGB探针只要有一个碱基突变,MGB探针就会检测到(MGB探针将不会与目的片段杂交,不产生荧光信号)。
因此,在进行SNP检测时,为了检测到突变子,即TaqmanMGB不与目的片段杂交,不产生荧光信号,探针目的片段产生荧光信号检测将探针的突变位点尽量放在中间1/3的地方。
注意:
为了满足上述要求的4个条件,探针的突变位点可向3’端移动,但突变位点至少在离3’端2个碱基的前方(即必须确保探针的后两个碱基是绝对的保守),以进行SNP检测。
反过来,若要进行同类检测,找的是保守片段区,探针中不应有突变位点。
若探针即便是只有13个bp,探针仍不完全保守。
有几个突变,突变位点也应靠近探针的5’端,这样,即便是突变,探针也可与目的片段杂交,产生荧光信号。
另一种方法是设计简并探针,也可达到即使是突变,仍可检测到突变。
第三步:
寻找一家信赖的公司合成引物和探针,一般引物合成大家比较熟悉,而且价格也比较便宜(特别是这两年便宜了许多),而探针则相对来说贵了许多,一般Taqman探针合成在1000到5000元不等(不同的合成要求价钱不同)——而这只是标记价钱,序列合成基本上和引物合成价钱相似。
第四、五、六步:
一般的定量PCR反应体系与普通PCR其实也差不了多少,只是要加入Taqman探针,另外不同就是分步法的不同。
其中需要注意的是:
*扩增酶最好选用热启动酶
*引物和探针的浓度需要进行优化,有人建议从50nM开始,在50nM—900nM之间优化,一般为200nM(注意探针需要避光保存。
*同样Mg+和酶量也需要进行优化,酶的推荐反应浓度是1.25-1.5U(50ul)
*DNA模板的添加量通常在100ng以下,因不同种类的DNA模板中含有的靶基因的拷贝数不同,必要时可进行梯度稀释,确定最佳的DNA模板添加量。
如果欲进行2StepRT-PCR反应的第二步PCR扩增反应,第一步的RT反应液作为DNA模板时的添加量不要超过PCR反应液总体积的10%。
另外循环参数虽然在引物和探针设计完之后也就确定了,但是有时也需要进行优化。
第七步:
在进行数据分析的时候,通常用不同浓度的标准样品的Ct值来产生标准曲线,然后计算相对方程式。
方程式的斜
度可以用来检查PCR的效率,对于100%PCR效率来说,一个理想的斜率是3.32。
最佳的标准曲线是建立在PCR的扩增效率为90%-100%(100%意味着在每个循环之后,模板的总数将增加为前一次的2倍)的基础上。
所有标准曲线的线性回归分析需要存在一个高相关系数(R2≥0.99),这样才能认为实验的过程和数据是可信的。
使用这个方程式我们可以计算出未知样本的初始模板量。
大多数定量PCR仪都有这样一个软件,它可以从标准曲线中自动地计算出未知样本的初始模板量。
这其中有两种基本的方法:
绝对定量和相对定量,研究人员需要根据自己的实验目的来选择。
绝对定量是指将未知样品与标准曲线相比较进行分析,一般标准品就是一个已知绝对浓度的DNA样品,要注意得是绝对定量分析的准确性是相对标准品的准确性而言的。
相对定量是指两个或更多的基因互相进行比较,其结果是一个比率,没有确切的数字被检测道。
另外由于不同的样品在反应过程中存在着一定的差异,因此除了要制作标准曲线来进行定量外,还需要设计表达水平相对较为稳定的内参基因来对结果进行标准化。
β-actin和三磷酸甘油醛脱氢酶(Glyceraldehyde-3-phosphatedehydrogenase,GAPDH)是两种较常用的管家基因,另外还有cyclophilin,18srRNA,phosphoglyserokinase,beta-microglobulin,beta-glucronidase,hypoxanthineribosyltransferase,transferringreceptor等。
要注意这些基因可以被反应条件所影响,在设计定量表达研究时,保证初始对照基因的质量是必要的一步,而且严谨的研究人员会通过对一系列内参基因定量结果取几何平均数来对定量数据进行标准化。
还有一个问题就是如何判断所得到数据的好坏,关于扩增曲线,经验总结认为:
“1.总体看曲线拐点清楚,指数期明显,扩增曲线整体平行性极好,基线平无上扬现象,低浓度样本扩增曲线指数期明显。
2.曲线指数期斜率,反应了扩增效率,越大说明扩增效率越高。
扩增效率越高,试剂灵敏度表现会越好。
3.基线,图上的基线也就是阴性样本的扩增曲线,平直或略微下降是好试剂的表现,阴阳性清楚,不易误判。
如果有上扬趋势,有可能造成阴性标本误判。
4.曲线与曲线间平行性非常好,说明各反应管扩增效率相近,外标准定量建立在每管扩增效率一样的假定基础上,所以扩增效率越相近,定量的重复性和准确性就越好。
而扩增效率相近与否,反应在扩增曲线图上就是曲线的平行性。
5.低浓度曲线指数期明显,一方面不易出现假阴阳性误判,另一方面说明灵敏度高。
”
实时荧光定量PCR实验体系的设计与优化
作者:
未知来源:
基因公司时间:
2007-8-1
时荧光定量PCR以其精确、快速、方便,越来越多的应用在科研、临床及检验检疫的各个领域。
但是定量PCR是对精确性要求很高的实验,不仅要求在实验前有比较完整的实验设计方案,而且实验的条件对实验结果的影响也非常大。
这些都是很多老师与学生非常关心的问题,下面分别从这两个方面来对定量PCR实验做一些阐述。
定量PCR实验步骤(以mRNA为例):
1.设计实验方案:
例如对样品、实验组和对照组的一个实验流程设计
2.引物和探针的设计和合成
3.抽提RNA,测定提取的RNA的浓度
4.反转录PCR
5.定量PCR
6.数据分析
在进行定量PCR实验的过程中,PCR的扩增效率是一个非常重要的影响因素,因为定量PCR原理的理论方程是基于扩增效率最大值1,因此高的扩增效率能保证定量PCR实验的精确性及重复性,影响PCR扩增效率主要有以下几个方面:
1,扩增子的长度;2,扩增子的GC含量;3,扩增子、引物和探针的二级结构;4,PCR反应各组分的浓度;5,RNA或者cDNA的纯度。
进行定量PCR实验时,必须设计好引物和探针,除了能获得高的扩增效率外,对PCR扩增的特异性、消除基因组DNA的扩增及提高扩增的灵敏度都有很大的影响。
下图就是使用不同的引物和探针对18SRNA进行定量PCR的荧光曲线图(反应条件和模板都相同)。
引物设计原则:
1.上下游引物要保守为了能够扩增出所需要的保守片段,必须对保守的100-200片段进行PCR扩增。
所以引物的选取也要非常的保守。
2.上下游引物的长度一般为18-30bp之间,且Tm值在58-62℃之间,上下游引物的Tm值相差最好不超过2℃。
3.确保引物中GC含量在30-80%。
应避免引物中多个重复的碱基出现,尤其是要避免4个或超过4个的G碱基出现。
引物的3’端最好不为G或/和C。
引物3’端的5个碱基不应出现2个G或/和C。
4.避免引物内出现反向重复序列形成发夹二级结构,同时也应避免引物间配对形成引物二聚体。
5.跨外显子设计引物,用于区别或消除基因组DNA的扩增。
探针设计的基本原则:
1.保守:
探针要绝对的保守,有时分型就仅仅依靠探针来决定。
理论上有一个碱基不配对,就可能检测不出来。
2.Taqman探针的长度最好在25-32bp之间,且Tm值在68-72℃之间,确保探针的Tm值要比引物的Tm值高出5-10℃,这样可保证探针在退火时先于引物与目的片段结合。
3.确保探针中GC含量在30-80%。
4.避免探针中多个重复的碱基出现,尤其是要避免4个或超过4个的G碱基
5.探针的5’端不能为G,因为即使单个G碱基与FAM荧光报告基团相连时,也可以淬灭FAM基团所发出的荧光信号,从而导致假阴性的出现。
6.Taqman探针应靠近上游引物,即Taqman探针应靠近与其在同一条链上的上游引物。
两者的距离最好是探针的5’端离上游引物的3’有一个碱基。
7.避免探针与引物之间形成二级结构。
8.对于多重定量PCR,例如SNP分型检测时,SNP位点应设计在探针的中间位置,并且两种探针的Tm值应相近。
实时定量PCR体系的优化
1.基本参数的优化:
1)MgCl2的浓度:
在PCR反应中,MgCl2的浓度对酶的活性是至关重要的,不仅如此,合适的MgCl2的浓度还能在反应中得
 升级会员
升级会员 