蛋白质工程重点内容总结Word格式.docx
《蛋白质工程重点内容总结Word格式.docx》由会员分享,可在线阅读,更多相关《蛋白质工程重点内容总结Word格式.docx(12页珍藏版)》请在冰豆网上搜索。
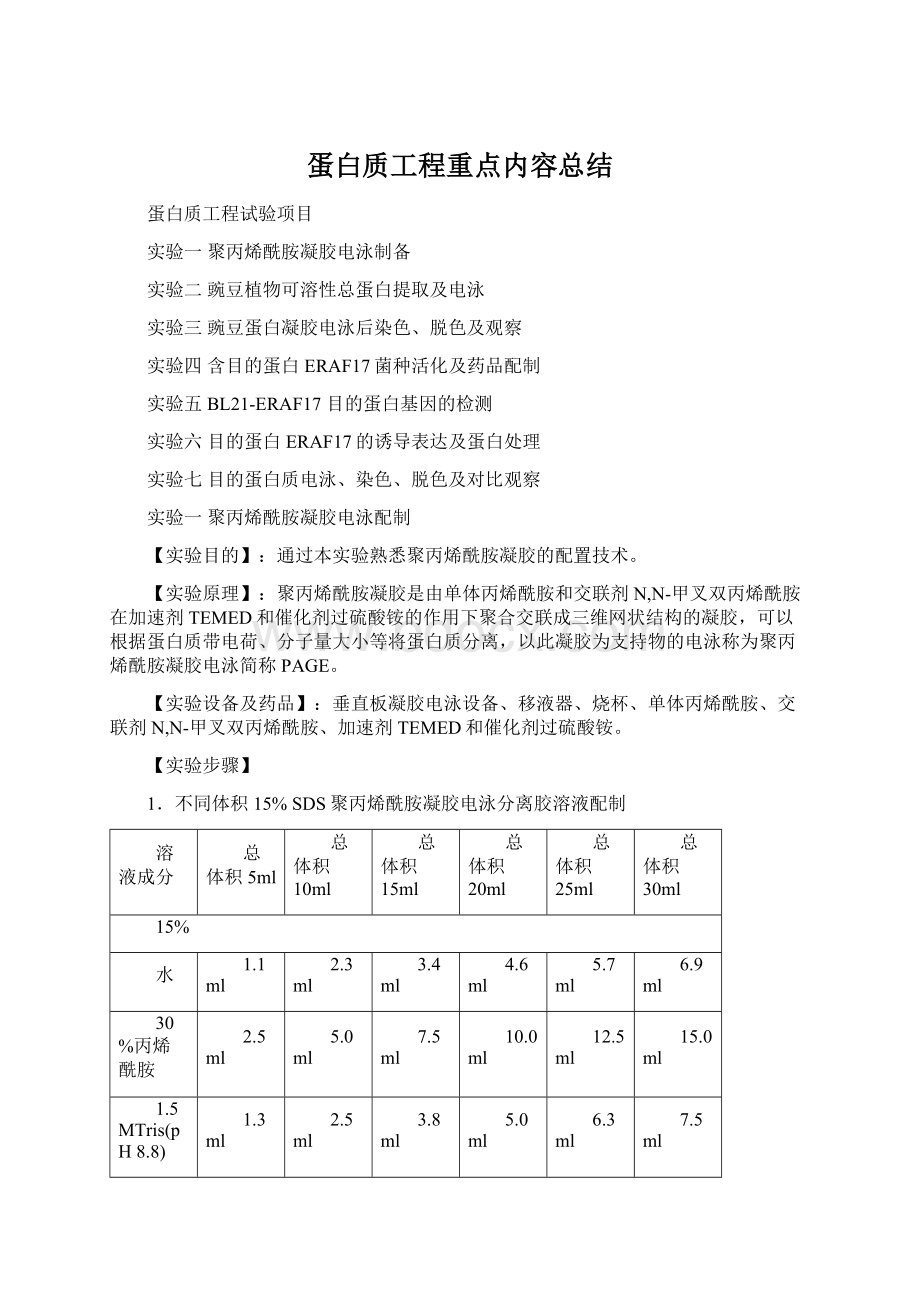
6.9ml
30%丙烯酰胺
2.5ml
5.0ml
7.5ml
10.0ml
12.5ml
15.0ml
1.5MTris(pH8.8)
1.3ml
3.8ml
6.3ml
10%SDS
0.05ml
0.1ml
0.15ml
0.2ml
0.25ml
0.3ml
10%过硫酸胺
TEMED
0.002ml
0.004ml
0.006ml
0.008ml
0.01ml
0.012ml
2.迅速在两玻璃板的间隙中灌注丙烯酰胺溶液,留出灌注浓缩胶所需空间(梳子的齿长再加0.5cm)。
再在胶液面上小心注入一层无水乙醇(异丙醇或水)(约2~3mm高),以阻止氧气进入凝胶溶液。
3.分离胶聚合完全后(约30分钟),倾出覆盖水层,再用滤纸吸净残留水。
4制备浓缩胶:
按下表给出的数据,在另一小烧杯中制备5%浓缩胶溶液4ml,一旦加入TEMED,马上开始聚合,故应立即快速旋动混合物并进入下步操作。
不同体积5%聚丙烯酰胺凝胶电泳浓缩胶制备
总体积3ml
总体积4ml
总体积6ml
总体积8ml
2.1ml
2.7ml
4.1ml
5.5ml
0.5ml
0.67ml
0.83ml
1.0ml
1MTris(pH6.8)
0.38ml
0.63ml
0.75ml
0.03ml
0.04ml
0.06ml
0.08ml
0.003ml
0.005ml
5.聚合的分离胶上直接灌注浓缩胶,立即在浓缩胶溶液中插入干净的梳子。
小心避免混入气泡,再加入浓缩胶溶液以充满梳子之间的空隙,将凝胶垂直放置于室温下。
学习植物可溶性总蛋白提取原理及方法,熟悉蛋白质电泳操作步骤,熟悉蛋白质分离的原理。
多数蛋白质都溶于水、盐溶液或有机溶液,分离纯化某一蛋白质首先要求把蛋白质从组织或细胞以溶解的形态释放出来,并保持原来的天然状态。
在变性聚丙烯酰胺凝胶电泳中由于SDS带有大量的负电荷,由于不同的SDS-蛋白质复合体具有相同的荷质比和相同的构象,因此迁移率不受原有的电荷、分子形状的影响,只取决于蛋白质分子量,因此经过一段时间电泳后蛋白质混合物会按其分子量大小分离。
高速离心机、移液器、蛋白质电泳仪、微量移液器、恒温水浴锅、Tris-甘氨酸电极缓冲液、液氮、豌豆苗。
【实验步骤】:
1.豌豆种植,8-10天后全株植物可作为实验材料提取可溶性蛋白
2.提取缓冲液(PBS)的配置
100ml
定容后调节pH值(7.4)。
3.将PBS溶液置于4度预冷。
4.将研钵,离心管,药匙用液氮预冷。
5.取各样品叶片分别约100mg,注意取材后要立即称量。
6.将叶片放入研钵,加入液氮进行充分研磨。
7.将研磨所得的粉末转移到Eppendorf管中,加入冰冷的PBS溶液200μl,颠倒混匀,立即置于冰上静止3-4小时(使其充分溶解)。
8.12000转/分在4℃条件下离心5分钟。
9.取上清液备用。
10.在蛋白质样品中加入1x的上样缓冲液,在100℃加热8-10分钟使蛋白质预变性。
11.待浓缩胶聚合完全后(30分钟),小心移出梳子。
把凝胶固定于电泳装置上,上下槽各加入Tris-甘氨酸电极缓冲液,必须设法排出凝胶底部两玻璃板之间的气泡。
12.按顺序加样,加样量通常为30μl(1.5mm厚的胶)。
13.将电泳装置与电源相接,凝胶上所加电压为8V/cm。
当染料前沿进入分离胶后,把电压提高到15V/cm,继续电泳直至溴酚蓝到达分离胶底部上方约1cm,然后关闭电源。
14.从电泳装置上卸下玻璃板,用刮勺撬开玻璃板。
紧靠最左边一孔(第一槽)凝胶下部切去一角以标注凝胶的方位。
了解蛋白染色脱色原理。
考马斯亮蓝R-250即三苯基甲烷每个分子含有两个SO3H基团,偏酸性,结合在蛋白质的碱性基团上,考马斯亮蓝染色具有很高的灵敏度,在聚丙烯酰胺凝胶中可以检测到0.1mg的蛋白质形成的染色带。
酸-甲醇溶液使蛋白质变性,固定在凝胶中,防止蛋白质在染色过程中在凝胶内扩散。
水平摇床、蛋白质染色液及脱色液。
1.蛋白质染色液配制
0.25g考马斯亮蓝R250
50ml甲醇
75ml冰醋酸用蒸馏水定溶到1000ml
2蛋白质脱色液配制
冰醋酸75ml
甲醇50ml
定容至1L
3.电泳结束后,小心地将蛋白质凝胶剥离浸泡在染色液中,
染色1~2小时或过夜。
4.染色结束后,取出凝胶浸泡在脱色液中,置于水平摇床上轻摇2~3小时,必要时更换脱色液,直至凝胶上的背景与蛋白质的对比度明显、蛋白质带清晰可见,背景显色很浅为止。
5..脱色结束后,取下凝胶进行照相。
掌握菌种活化过程,熟悉无菌操作过程
菌种活化就是逐级扩大培养,是为了得到纯而壮的培养物,即获得活力旺盛的、接种数量足够的培养物。
菌种发酵有一般需要2-3代的复壮过程,因为保存时的条件往往和培养时的条件不相同,所以要活化,让菌种逐渐适应培养环境。
【实验设备及药品】无菌操净台、高压灭菌锅、移液器、LB液体及固体培养基、含有目的蛋白的菌种。
【操作步骤】:
1.实验准备阶段:
玻璃平板、50ml、150ml或200ml三角瓶,黄色及蓝色枪头。
2.LB培养基的配制:
液体:
1000ml
胰蛋白胨(Tryptone)10g
酵母提取物(yeastextract)5g
NaCl5g
PH=7
固体培养基,上述物质溶解后加入琼脂粉15g
121度高温灭菌20分钟,待固体培养基不是特别热的时候加入卡那霉素,使其终浓度为50mg/l。
3.取存于-70oC的菌种在固体LB培养基上划线,37oC温箱培养12-16h。
实验五兔源蛋白磷酸酶基因的检测
【实验目的】熟悉原核表达载体中目的片段与表达蛋白所对应的关系,对原核表达载体目的基因进行鉴定。
【实验原理】对原核表达载体中目的基因进行PCR鉴定,通过PCR检测确定目的蛋白基因的存在与片段大小。
【实验仪器及药品】:
PCR仪、电泳仪、移液器、Taq聚合酶、紫外凝胶成像系统。
一、提取质粒
1.收集菌体2ml,13000rpm,离心2min,弃上清液。
2.加入150μlSolⅠ(含Tris,EDTA,葡萄糖)重悬菌体,可用枪头剧烈振荡。
3.加入150μlSolⅡ(0.2MNaOH,,1%SDS(m/v),需要现用现配,将0.4MNaOH溶液和2%SDS溶液等体积混合),小心轻混,缓慢上下颠倒2-3次。
此时溶液呈黏稠状。
4.加入150μlSolⅢ(冰醋酸,醋酸钾)小心轻混,缓慢上下颠倒2-3次。
有白色絮转沉淀。
5.13000rpm,离心3min。
,取上清液。
加入等体积(大约450μl)酚/氯仿/异戊醇(使用时要注意用枪头吸下液面部分)振荡混合。
此时溶液呈乳浊状。
6.13000rpm,离心3min,取上清液。
加入2倍体积无水乙醇(约900μl或1ml),置于-20oC醇沉10min。
7.13000rpm,离心3min,弃乙醇。
加入1ml70%乙醇,13000rpm,离心3min,弃掉溶液,自然干燥15min。
注意保持质粒的湿润。
8.加入30μl灭菌水溶解质粒后,于4oC保存。
二、1%琼脂糖凝胶电泳的制备
1.选好胶板插好梳子。
2.称取0.25g琼脂糖溶于25mlTAE缓冲液中。
3.在微波炉中80火力溶解,溶解后冷却,不烫手时加入1.5ul的EBr倒入胶板中赶走气泡,使其凝固。
4.取2ul质粒加入2ulloadingbuffer混匀,上样。
三、PCR及电泳
总体系25ul
体系组分
用量(μl)
PremixrTaq酶
12.5
引物1(10μM)
1
引物2(10μM)
DNA模板
3
加ddH2O到25μl
1.PCR程序设定
(1)94℃预变性3分钟
(2)94℃变性50秒
(3)50℃退火50秒
(4)72℃延伸50秒
第二步到第四步30个循环
(5)72℃8分钟
(6)4℃保存
2.PCR产物进行琼脂糖凝胶电泳
1)选好胶板插好梳子。
2)称取0.3g琼脂糖溶于30mlTAE缓冲液中。
3)在微波炉中80火力溶解,溶解后冷却,不烫手时加入1.5ul的EBr倒入胶板中赶走气泡,使其凝固。
4)在PCR产物中加入5ulloadingbuffer后混匀。
5)选择DNA2000marker为标准上样。
6)PCR产物上样。
7)在紫外凝胶成像系统中观察实验结果。
实验六目的蛋白ERAF17的诱导表达
熟悉细菌液体培养过程,掌握诱导原核生物表达外源蛋白的原理及方法
目的基因ERAF17克隆在PET-28载体上,控制该基因的启动子为诱导型启动子需要在培养基中加入乳糖类似物IPTG目的基因才会表达,加入IPTG一定时间后目的蛋白在细胞中不断的积累,一定时间后表达量最大。
37度恒温摇床、移液器、恒温水浴锅、IPTG。
1.挑取含有目的基因ERAF17的单菌落,接种于5mlLB培养液中(含50µ
g/ml,卡那霉素),37℃振荡培养过夜
2.取过夜培养液按1:
100的比例扩大培养。
3.在OD600值0.6-0.7时取1ml菌液收集菌体备用。
取完菌液后,在培养液中加入IPTG至终浓度1.0mmol/L进行诱导,经不同的诱导时间(1h,2h,3h,)后取1ml培养物收集菌体。
4.将收集的所有菌体用1×
蛋白上样缓冲液重悬,100℃8-10min后离心,取上清备用。
进一步熟悉聚丙烯酰胺凝胶电泳的配制方法及电泳过程,学习对比观察电泳结果。
目的基因在IPTG的诱导下表达,在不同时期收集大肠杆菌表达的蛋白质,通过电泳后会看到目的蛋白,没有经过诱导的大肠杆菌不表达该蛋白因此经过电泳后没有目的带。
蛋白质垂直电泳仪、移液器、蛋白染色及脱色液。
1.聚丙烯酰胺凝胶电泳配制
方法见实验二(详细步骤)
2.取原核生物大肠杆菌诱导前、后各阶段的蛋白质进行电泳。
泳道1低分子量蛋白Marker,按收集先后顺序进行电泳。
3.电泳后进行染色(过夜)。
4.脱色及脱色。
5.对比观察。
6.拍照保存实验结果。
蛋白质工程备选实验
综合性实验目的蛋白原核表达载体的构建
实验一、目的蛋白基因片段的克隆(6学时)
【实验目的】学习植物RNA的提取,通过反转录PCR从植物基因组中克隆目的蛋白基因。
【实验原理】根据中心法则,信使RNA翻译成蛋白质或多肽片段,欲获得在原核生物中可表达的蛋白质需首先获得该蛋白质的基因片段,使用植物mRNA提取试剂盒提取总mRNA,通过反转录PCR可获得cDNA,针对目的基因设计引物克隆目的基因。
低温高速离心机、制冰机、移液器、1.5ml离心管、植物RNA提取试剂盒。
一、植物RNA提取
1.在液氮中研磨100mg植物叶片,将粉末加入到1.0ml的Trizol。
2.用1ml针筒,26-G号(6#)针头抽吸匀浆液以剪切基因组DNA,然后直接从针筒中将样品转移到无菌1.5ml离心管中。
3.加入200μl氯仿/异戊醇(24:
1)或氯仿,剧烈振荡混匀30秒。
4.台式离心机上,12000rpm,室温离心5分钟。
5.将上清液小心转移到Rnase-free1.5ml离心管中,加入等体积的异丙醇,室温下放置5分钟。
6.台式离心机上,12000rpm,室温离心2分钟。
7.尽可能彻底地吸走上清,防止RNA沉淀丢失。
8.真空离心干燥3-5分钟,或放在室温下使酒精完全挥发。
9.沉淀用30-50μlDEPC-H2O溶解。
如发现沉淀难溶,68℃处理10分钟。
对于胰腺,肾等组织中Rnase含量很高,沉淀用100%去离子甲酰胺溶解。
二、反转录PCR
1.用不含RNA酶的DNA酶处理制备的RNA以除去可能含有的DNA杂质。
2.根据RT-PCR试剂盒所提供的操作手册进行反转录及PCR反应。
20μl反转录体系如下:
体积单位μl
MgCl2
10xbuffer
dNTP
Inhi-bitor
反转录酶
引物
模板RNA
H2O
检测样品
4
2
0.5
7
注:
1.本反应体系中的引物为Oligo(dT)15Primer。
2.转录条件:
42℃1小时;
95℃5分钟;
4℃5分钟。
3.PCR反应:
将管中的RT反应液稀释至100μl,取10μl为模板进行PCR扩增。
三、通过特定引物克隆目的基因片段
反应体系为:
25ul体系
实验二、目的基因片段回收并与载体连接(6学时)
将目的基因片段与原核表达载体连接。
目的片段与目的载体同时使用相同的酶进行酶切处理,将两者按一定比例混合后可连接为新的重组子。
37度恒温培养箱、制冰机、移液器、连接酶、DNA片段回收试剂盒。
1.目的片段PCR产物电泳
2.电泳产物回收
1).切去含DNA片段的琼脂糖(100-300mg),尽量切得小一点,用吸头捣碎按质量比1:
3(切取重量:
溶液A的体积比)加入溶液A。
2).55-65度水浴10min,直至胶完全溶化,期间可振荡助溶2-3次,待溶化后置室温加入50ul溶液B,充分混匀。
3).将溶液置于离心柱中,静置2min,≥8000转离心30秒,若一次加不完,可分两次离心。
(静置2min,是必须的,否则明显影响回收效率).
4).倒掉液体,加入500ul溶液C于离心柱中8000转离心30秒,弃溶液。
5).重复步骤4。
6).12000转再次离心1min,甩干剩余液体以除去残余酒精.
7).将离心柱置于新的离心管中,温室敞开离心管盖放置5-10min,使乙醇挥发殆尽。
8).加入20-30ul溶液D(50度水浴),静置2分钟。
9).12000转离心2分钟,管底溶液即为所需DNA。
将DNA储存于-20度可长期保存。
3.目的片段与载体双酶切
4.酶切产物回收后连接反应体系如下20ul
目的片段10ul
载体片段3ul
连接酶1ul
连接buffer2ul
水4ul
实验三连接产物转化及鉴定(6学时)
通过本实验学习重组子扩增的原理及鉴定。
当细菌处于容易吸收外源DNA的状态时,为感受态,低温时质粒容易与感受态细胞吸附,经升温热刺激后被感受态细胞吸收,质粒上有编码相应抗性的基因,因此转化的细菌在抗性平板上生长。
37度恒温培养箱、37度恒温振荡摇床、无菌操净台。
一、连接产物转化
1.将所有连接反应体系加入到200μlDH5a感受态细胞中,轻轻旋转,混匀内容物,冰浴30min。
2.将1.5μlEP管放入预加热的42oC水浴锅中,恰恰放置90s,热休克时不可摇动EP管。
3.迅速将EP管移到冰中,使细胞立即冷却2min。
4.加入200μLB培养液,37oC(160-225)rpm摇床培养1h。
5.将管中液体全部取出涂布于含相应抗性的LB培养基平板上(平板上加入卡那抗生素50mg/ml)37oC温箱培养12-16h。
6.次日,观察是否有菌落长出。
二、重组子鉴定
一、PCR过程
1.在5mlLB培养基中加入终浓度为50mg/l的卡那霉素。
2.挑取单菌落于LB培养基中37度摇床过夜培养。
3.提取重组子质粒(提取操作见实验二)进行PCR鉴定。
4.取模板2-3ul加入装有预混Taq聚合酶的pcr小管中。
体系如下:
25ul体系
5.将样品混合物混匀置于PCR仪中。
二、PCR产物进行琼脂糖凝胶电泳
1.选好胶板插好梳子
2.称取0.3g琼脂糖溶于30mlTAE缓冲液中
3.在微波炉中60火力溶解,溶解后冷却,不烫手时加入1.5ul的EBr倒入胶板中赶走气泡,使其凝固
4.在PCR产物中加入5ulloadingbuffer后混匀
5.选择DNA2000marker为标准上样
6.PCR产物上样
7.在紫外凝胶成像系统中观察实验结果。
 升级会员
升级会员 