GaussView高级技巧.docx
《GaussView高级技巧.docx》由会员分享,可在线阅读,更多相关《GaussView高级技巧.docx(9页珍藏版)》请在冰豆网上搜索。
GaussView高级技巧
GaussView高级技巧
(一):
十个基本高级技巧
摘要
本文主要介绍了利用GaussView分子模型的十个高级技巧,其中大多数技巧是鲜为人知的。
这些技巧的组合能够使GaussView发挥巨大威力,可以快速方便地构造出复杂的分子结构。
最后,以构造乙烷的重叠式构象和二茂铁结构为例进一步说明了这些高级技巧的具体应用。
本文主要介绍利用GaussView构建分子模型的高级技巧,希望对大家有所帮助。
本文所使用的版本是GaussView3。
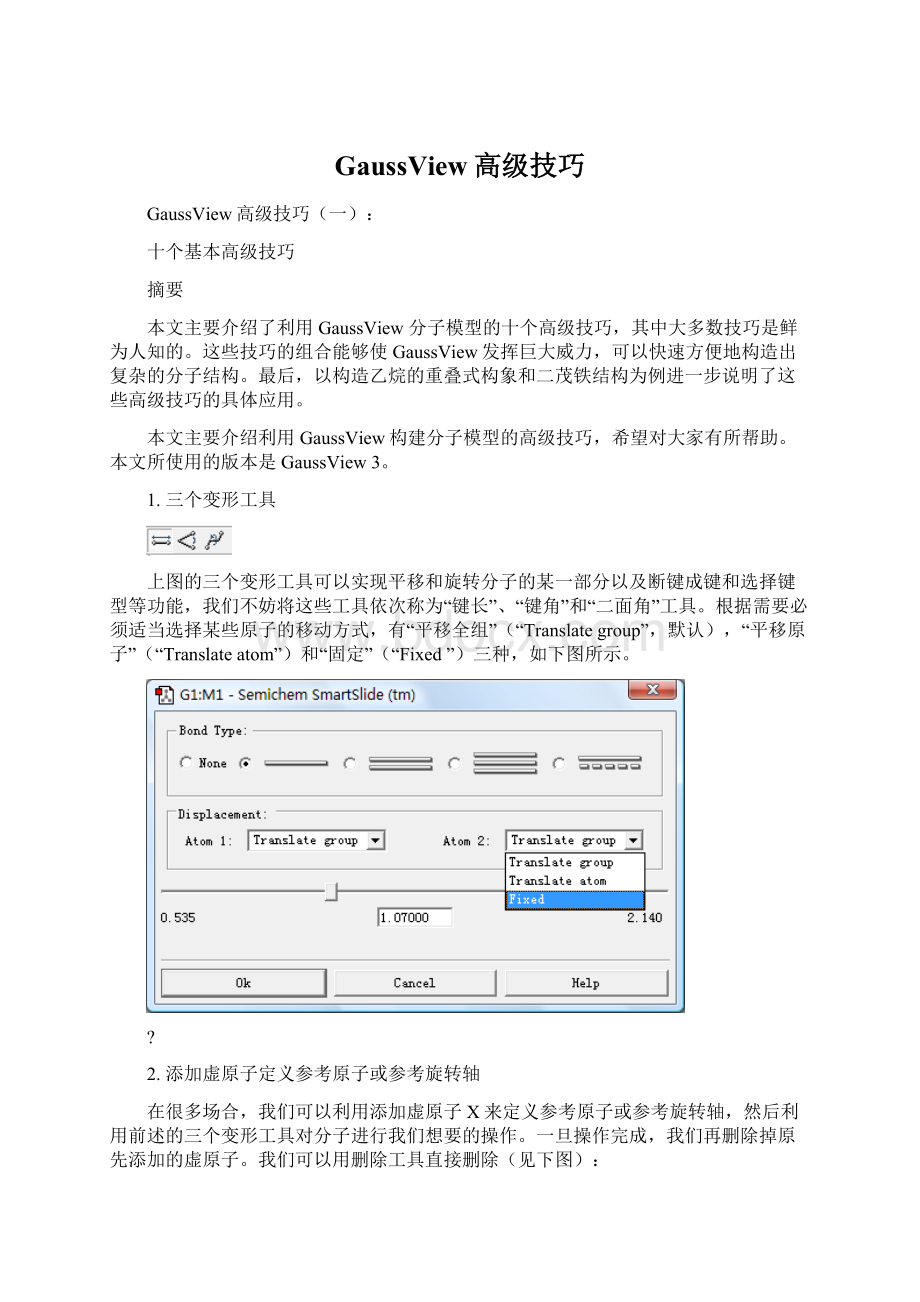
1.三个变形工具
上图的三个变形工具可以实现平移和旋转分子的某一部分以及断键成键和选择键型等功能,我们不妨将这些工具依次称为“键长”、“键角”和“二面角”工具。
根据需要必须适当选择某些原子的移动方式,有“平移全组”(“Translategroup”,默认),“平移原子”(“Translateatom”)和“固定”(“Fixed”)三种,如下图所示。
?
2.添加虚原子定义参考原子或参考旋转轴
在很多场合,我们可以利用添加虚原子X来定义参考原子或参考旋转轴,然后利用前述的三个变形工具对分子进行我们想要的操作。
一旦操作完成,我们再删除掉原先添加的虚原子。
我们可以用删除工具直接删除(见下图):
但由于虚原子体积很小,有时会被其他原子覆盖,这时无法用删除工具直接删除。
更通用的删除方法是:
点击“原子列表”按钮(也可以选择菜单Edit>AtomList...),见下图。
在弹出的“AtomListEditor”窗口中选中要删除的原子,然后选择菜单Edit>DeleteAtoms>Selected。
3.利用加氢工具获得最佳成键位置
利用上图的加氢工具可以对某个原子添加一个氢原子。
但这个氢原子的成键取向有时并非我们想要的。
解决办法是:
既然我们第一个加氢的取向不理想,那么我们就再添加一个,就这样连续不断地加氢,直到所加的氢原子的位置符合我们需要位置。
然后利用删除工具将前面添加的氢原子全部删除即可。
加氢工具的另一个目的就是为后面添加其他基团做准备。
用下面介绍的基团工具点击氢原子将氢原子取代掉后即可达到在一定的成键方向添加某个基团的目的。
4.基团取代工具
如上图所示,基团取代工具有:
元素基团、环状基团、R基团、生物基团和自定义基团。
元素基团可以添加或取代各种杂化状态的原子基团,其余各种基团工具则提供许多常用基团的模板,在此基础上我们可以构建相当复杂的分子。
我们可以点击当前基团(“CurrentFragment”)面板中的“Hot”位置来选择取代位置,如下图所示。
5.点群工具
选择菜单Edit>PointGroup...即弹出上图的点群工具对话框,选定适当的容差值即可将分子进行对称化,使分子具有某种点群对称性。
点击“Symmetrize”按钮后,分子就已经具有相应点群对称性了。
如果再选择“Constraintosubgroup”中的某个点群,则笛卡尔坐标轴的定向将按照分子的对称轴进行选取,如下图所示。
如果再选中下方的“Alwaystrackpointgroupsymmetry”选项,则后面当我们利用变形工具改变某个几何参数(键长或键角或二面角)时或者删除工具删除某个原子时,分子将按照对称性的要求自动改变其他对称相关的几何参数(键长或键角或二面角)或删除其他对称相关的原子。
一旦分子具有某种点群对称性后,坐标轴原点将平移到分子的几何中心,而且分子的主轴被取为z轴。
可见,有时适当地使用点群工具能事半功倍。
6.利用晶体工具在一定方向复制分子
晶体工具也可以通过选择菜单Edit>PBC...调出。
晶体工具除了构造一维至三维晶体结构外,还可以实现分子的平移复制。
方法是:
在弹出的PBC对话框中选择Symmetry选项卡中的“LatticeDimension”中的“Three”,即构造三维晶体结构(一般来说三个晶轴的方向中的一个是沿着分子主轴的方向)。
我们可以通过调节Alpha至Gamma角来得到我们想要的一个晶轴取向,然后在“Cell”选项卡中设定晶格长度(a、b和c的大小)。
再在“View”选项卡中选择该晶轴方向复制的数目,最后点击“Combine”按钮既可。
完成后可以将PBC对话框中Symmetry选项卡的“LatticeDimension”一项设为“None”,即可取消周期性条件。
当然,我们也可以使用复制和粘贴工具进行分子的复制和粘贴,如下图所示:
但其局限性在于只能按照软件默认的方向粘贴分子,因此往往需要一些后续操作才能符合我们的要求。
7.合并原子
有时我们需要合并两个原子,以达到连接两基团的目的。
通过键长工具将两原子的键长设定为?
(这是键长设定的最小值),然后用删除工具删掉其中一个原子就可以将两原子合并。
有时我们需要先将某一个原子换成稍大的不同颜色的原子,这样合并后便于使用鼠标准确地删除。
8.平移分子的部分基团
首先需要通过键长工具进行适当的断键和成键,从而确定待旋转的基团。
然后利用键长工具分别点击待平移基团的关键原子和位置参考原子,并设定Atom1为“Translategroup”,Atom2为“Fixed”,再调节键长大小既可实现平移。
必要时需要添加虚原子来定位。
9.旋转分子的部分基团
首先需要通过键长工具进行适当的断键和成键,从而确定待旋转的基团。
然后利用二面角工具(有时也可能用上键角工具)分别点击待平移基团的某些关键原子和位置参考原子,并设定Atom1为“Translategroup”,Atom4为“Fixed”,再调节二面角大小既可实现旋转。
必要时需要添加虚原子来定义旋转轴。
10.显示坐标轴可以更准确地添加原子
点击右键菜单可以选择显示笛卡尔坐标轴。
显示坐标轴可以更准确地添加原子,比如将原子放在z轴的方向上。
此外,值得注意的是,任何被添加的原子总是处在垂直屏幕方向的分子纵深长度的中心。
下面举几个实例来具体说明上述技巧的使用。
1.构造乙烷的重叠式构象
用GaussView直接构造乙烷的交错式构象很容易:
先用元素基团工具中的sp3杂化的C按钮点击新建窗口便得到甲烷分子,然后在点击其中一个氢原子即得到乙烷的交错式构象(技巧4)。
在此基础上我们来构造乙烷的重叠式构象。
思路很简单,只需将其中一个甲基相对于另一个甲基绕C-C键旋转60度即可。
?
用二面角工具按上图利选择1到4号原子,按照前述“旋转分子的部分基团”的技巧,将二面角从180度设为120度,就可以将右边的甲基旋转60度,从而得到乙烷的重叠式构象(技巧9)。
?
2.构造二茂铁结构
首先我们构造环戊烯结构。
利用环状基团工具中的环戊二烯基团按钮(GaussView没有环戊烯基团模型)点击空窗口,构造出环戊二烯(技巧4)。
用删除工具删除CH2的两个氢原子。
再用加氢工具加一个氢原子即可得到正确取向的氢(技巧3)。
用点群工具将该结构对称化为D5h点群(容差值为),同时选中“Alwaystrackpointgroupsymmetry”。
这样用键长工具将任一个C-C键长设为?
,键型设为“共轭双键”,则其余四个C-C键都拥有同等键长和键型。
同时具有D5h点群对称性的环戊烯分子中心就是坐标原点(技巧5)。
显示坐标轴,用鼠标操作使分子的xy平面大致处于屏幕平面,然后在坐标中心的位置点击添加一个虚原子(技巧10),如上图所示。
这个虚原子后面有用。
下面我们构造另一个平行的环戊烯分子。
打开晶体工具,选择c方向的晶格长度为?
,并设c方向的重复单元为2,点“Combine”合并,便得到如下图所示的两个相距?
且平行的环戊烯结构(技巧6)。
取消掉周期性条件。
但此时可以看到坐标轴原点不在两分子的中点位置,这样不利于添加铁原子以及后续操作。
为此,我们打开点群工具,将分子点群对称化为D5h。
,这样坐标轴原点就恰好在两分子的中点位置(技巧5)。
由于二茂铁中两个环戊烯是交错的构象,所以我们可以利用例1中的方法将其中一个环戊烯绕分子主轴旋转36度既可(但事先必须打开点群工具将“Constraintosubgroup”设为C1,否则D5h的对称性限制使得该旋转操作无法进行),如下图所示(技巧9)。
接着用鼠标操作使分子的yz平面大致处于屏幕平面,在坐标中心的位置点击添加一个铁原子(技巧10)。
用删除工具删除两个虚原子,但我们会看到残余的十个键(原先的C-X键)。
当然我们用删除工具手工一个个删除。
更简单的方法是,点击“重新键连”(“Rebond”)按钮(或选择菜单Edit>Rebond)即可消除那些残键。
最后用点群工具将整个分子对称化。
但我们会发现尽管GaussView识别分子属于D5d点群,但无论怎么点击“Symmetrize”按钮,分子仍然保持C1点群。
其原因是模型中仍有一些残键,这些残键是我们在使用晶体工具时留下的,因为当时的晶胞较小,相邻晶胞内原子自动成键。
因此我们需要打开原子列表窗口,选中那些残键(元素符号是“”的那些),然后删除即可。
此时再对称化就能得到D5h的二茂铁结构(技巧5)。
?
 升级会员
升级会员 