制药工程专业药理学考试重点总结.docx
《制药工程专业药理学考试重点总结.docx》由会员分享,可在线阅读,更多相关《制药工程专业药理学考试重点总结.docx(23页珍藏版)》请在冰豆网上搜索。
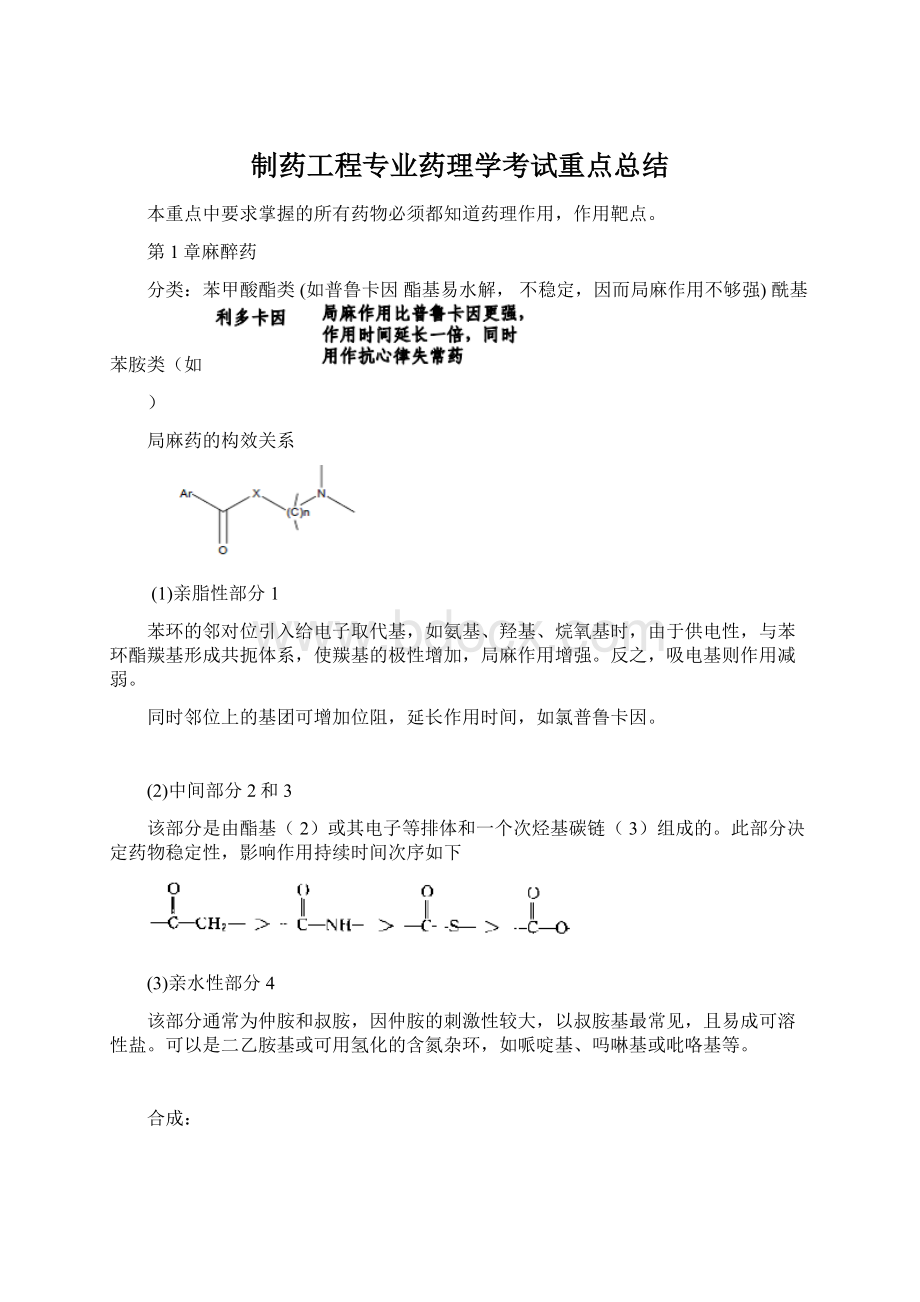
制药工程专业药理学考试重点总结
本重点中要求掌握的所有药物必须都知道药理作用,作用靶点。
第1章麻醉药
分类:
苯甲酸酯类(如普鲁卡因酯基易水解,不稳定,因而局麻作用不够强)酰基苯胺类(如
)
局麻药的构效关系
(1)亲脂性部分1
苯环的邻对位引入给电子取代基,如氨基、羟基、烷氧基时,由于供电性,与苯环酯羰基形成共扼体系,使羰基的极性增加,局麻作用增强。
反之,吸电基则作用减弱。
同时邻位上的基团可增加位阻,延长作用时间,如氯普鲁卡因。
(2)中间部分2和3
该部分是由酯基
(2)或其电子等排体和一个次烃基碳链(3)组成的。
此部分决定药物稳定性,影响作用持续时间次序如下
(3)亲水性部分4
该部分通常为仲胺和叔胺,因仲胺的刺激性较大,以叔胺基最常见,且易成可溶性盐。
可以是二乙胺基或可用氢化的含氮杂环,如哌啶基、吗啉基或吡咯基等。
合成:
利多卡因(作用于外周神经组织)
罗哌卡因(作用于外周神经组织)
第2章催眠镇静药、抗癫痫药和精神神经疾病治疗药
分类:
巴比妥类(苯巴比妥、异戊巴比妥、硫喷妥、司可巴比)
苯并二氮卓类(地西泮、硝西泮、奥沙西泮、艾司唑伦)等。
2.1巴比妥类催眠镇药
构效关系:
5位无取代和5位单取代均无作用(pKa过小);R1或R2为支链或不饱和烃基,作用时间短;若为饱和烃基、芳烃基,作用时间长(代谢有关);R1和R2总碳数为4~8,若>8作用相反或无作用(logP)。
R3为甲基,作用增强(降低解离度,增加脂溶性);若两个N上均有甲基取代即具有反作用(惊厥)。
X=O,S;S取代时脂溶性增加,起效快,作用时间短,X=NH无作用。
2.2苯二氮卓类催眠镇药
苯二氮卓类药物的构效关系
4,5位拼合四氢噁唑环,稳定性增加,作用强。
1,2位拼合三唑环稳定性增加,作用强。
1,4-二氮卓环为必须结构
苯二氮卓类的稳定性
(4,5位,1,2位在酸性及胃肠道中的开环反应)
2.4抗癫痫药
分类:
酰脲类(巴比妥类(如苯巴比妥)、乙内酰脲类(如三甲双酮,乙琥胺)),GABA类似物(GABA为中枢抑制性递质,如卤加比)。
2.4.1酰脲类
乙内酰脲的结构改造方法:
将乙内酰脲化学结构中的-NH-以其电子等排体-O-或-CH2-取代,分别则得到了噁唑烷酮类和丁二酰亚胺类。
2.4.2、GABA类似物
机制:
GABA为中枢抑制性递质,该类药物具有与GABA的类似结构,可通过抑制GABA氨基转移酶的活性,或为GABA的前药
代谢反应:
卤加比的体内代谢
2.5精神神经疾病治疗药
分类:
吩噻嗪类(氯丙嗪、奋乃静、氟奋乃静)
噻吨类(硫杂蒽类)(如氯普噻吨(泰尔登))
丁酰苯类(如氟哌啶醇、五氟利多)等
2.5.1抗精神病药(都是多巴胺受体阻断剂)
吩噻嗪类构效:
含N环上侧链部分必须由三个成直链的碳原子组成,若为支链,抗精神病活性明显下降,抗组胺作用增强。
顺式吩噻嗪类药物与多巴胺的优势构象能部分重叠,活性高
2位R3为吸电基,活性强。
NR1R2碱性基团常为叔胺,可为脂肪叔胺如二甲氨基,也可为氮杂环,常用哌啶基或哌嗪基,以哌嗪最强
合成:
氯丙嗪
抗精神病药的共同副作用:
锥体外系副作用
2.5.2抗抑郁药
NA和5-HT相对增多,表现为躁狂;NA和5-HT相对缺乏,表现为抑郁。
目前临床应用的抗抑郁药可分为去甲肾上酰素重摄取抑制剂(三环类抗抑郁药)、单胺氧化酶抑制剂及5—羟色胺再摄取抑制剂。
第3章解热镇痛药及非甾体抗炎药
解热镇痛药及NSAIDs的作用靶点主要是花生四烯酸环氧合酶(COX)。
通过抑制COX来阻断或减少前列腺素(PG)的合成。
另外部分还抑制脂氧化酶,减少白三烯等的合成。
COX-1促进生理性PGs的合成,调节正常组织细胞的生理活动。
如保护消化道粘膜,调节血小板功能。
COX-2在炎症细胞中高度表达,使炎症组织中前列腺素合成增加。
促使炎症进一步发展。
分类:
乙酰苯胺类(扑热息痛、非那西丁)
水杨酸类(阿司匹林、贝诺酯、阿司匹林铝、水杨酰胺)
芳基烷酸类(吲哚美辛、舒林酸;托美丁;依托度酸
;双氯芬那酸钠;布洛芬、萘普生、酮洛芬、氟比洛
芬)
协同前药(mutualprodrug):
贝诺酯(COXI,用于解热镇痛及抗炎,口服对胃无刺激,适合
老人和儿童使用。
)
为了减小阿司匹林的副作用,将阿司匹林和扑热息
痛通过酯键相连。
口服后在体内分解又重新生成原来的两个药物,
共同发挥解热镇痛作用。
采用前药原理和拼合原理,叫做协同前药
(mutualprodrug)
前药:
芬布芬在体内代谢成联苯乙酸的活性形式发挥药理作用。
(是酮酸类的前体药物,在体内代谢生成联苯乙酸而发挥作用。
联苯乙酸是COXI,口服芬布芬可避免直接服用联苯乙酸对胃肠道的刺激,胃肠道反应较小。
临床上用于类风湿性关节炎、风湿性关节炎,也可用于牙痛,手术后疼痛)
合成:
双氯灭痛
布洛芬
芳基烷酸的基本结构:
第4章镇痛药
(镇痛药镇痛作用部位主要在中枢,解热镇痛抗炎药作用部位一般认为是在外周神经)
有证据表明κ受体对μ受体介导的反应有调节作用,故而混合激动-拮抗剂是发展的方向,即κ受体激动剂及μ受体拮抗剂,用作镇痛药时一般成瘾性小,如喷他佐辛(μ受体弱拮抗剂,κ受体激动剂,首个用于临床的非麻醉性阿片类合成镇痛
药,镇痛效果为吗啡的1/3,优点是副作用小,成瘾性低。
左旋体的活性是右旋
体的20倍,临床上应用其消旋体)为κ受体激动剂及μ受体弱拮抗剂。
μ受体镇痛活性最强,成瘾性也最强
δ受体成瘾性小,镇痛作用也不明显
(该章药物作用特点主要回答有镇痛作用强弱和有无成瘾性。
)
4.1吗啡及其结构改造
吗啡的性质:
具有酸碱两性,3位弱酸性的酚羟基,17位碱性N-甲基叔胺
纳洛酮的作用特点:
阿片μ受体拮抗剂,用于吗啡中毒解救
吗啡的构效关系(半合成类镇痛药)
(吗啡)
叔胺是镇痛活性的关键基团,氮原子引入不同的取代基可使μ受体激动剂转变为拮抗剂。
酚羟基被醚化和酰化后,活性及成瘾性均降低。
羟基被烃化、酯化、氧化或去除后,活性及成瘾性均增加。
4.2全合成镇痛药
分类:
吗啡烃类(N-甲基吗啡烃,左啡诺)
苯并吗喃类(喷他佐辛等)
苯基哌啶类(哌替啶)
苯基丙胺类/氨基酮类(盐酸美沙酮)
合成:
哌替啶
盐酸美沙酮的作用特点:
μ受体激动剂,成瘾性较吗啡小,成瘾发生慢。
用作戒毒药。
有效镇痛药的结构要求,或构效关系如下:
第5章心血管系统药物
5.1强心药
强心药主要增强心脏收缩力,临床常用药主要有:
强心苷类、β-受体激动剂
强心苷类抑制心肌细胞膜结合的Na+,K+-ATP酶(钠泵),并进一步导致细胞内参与激活心脏收缩力的Ca2+增加。
5.2作用于离子通道药物
当前作用于离子通道的心血管药物主要有钙离子通道阻滞剂、钠离子通道阻滞剂、钾离子通道阻滞剂、钾离子通道开放剂。
5.2.1钙通道阻滞剂/钙拮抗剂
钙拮抗剂的作用与临床应用:
抗心绞痛抗心率失常抗高血压
钙拮抗剂的结构类型:
二氢吡啶类等
基本结构:
合成:
硝苯地平
稳定性:
硝苯地平长期暴露在空气中或在紫外光照射下反应产物
钠通道阻滞剂作用:
抗心律失常(具有抑制钠离子内流和促进钾离子外流的作
用,用于急、慢性室性心律失常)。
作用特点:
利多卡因的两个药理作用(降低去极化最大通量,缩短动作电位时间)
5.2.3钾通道阻滞剂
钾通道作为III类抗心律失常药。
5.2.4钾通道开放剂
钾通道开放剂临床用于治疗高血压等。
5.3作用于肾上腺素能神经系统的药物
主要有Mg2+-ATP酶抑制剂(胺泵抑制剂)、作用于α受体药物、β受体拮抗剂。
5.3.1Mg2+-ATP酶抑制剂
作用特点:
该类药物多具有降压作用缓慢、温和而持久的特点。
稳定性:
碱性条件下的水解
5.3.2作用于肾上腺素能受体的药物
主要有α2受体激动剂、α1受体阻滞剂和β受体阻滞剂。
常用的β受体阻滞剂按作用的选择性分类分为三种:
非选择性β受体阻滞剂、选择性β1受体阻滞剂、非典型β受体阻滞剂。
β受体阻滞剂的基本结构和构效关系
β碳为手性C,苯乙醇胺型,R>S,芳氧丙醇胺型,Rβ碳上羟基为活性必须基团,被取代或醚化作用消失,酯化可延长时间。
n=0,苯乙醇胺型合成:
可乐定
5.4影响肾素-血管紧张素-醛固酮系统的药物
5.4.2血管紧张素转化酶抑制剂(ACEI)
此类药物有三类:
含巯基类(卡托普利)、含二羧基类(依那普利)、含磷酰基类(福辛普利)
合成:
卡托普利
ACE抑制剂的构效关系:
(1)ACEI分子中有三个基团与ACE的结合点相结合:
与ACE的锌离子结合的基团,如巯基、羧基、次磷酰基,与ACE的正电荷以离子键结合的阴离子部分,即末端氨基酸部分的羧基,与ACE的供氢部位以氢键结合的基团,即酰胺部分的羰基
5.4.3血管紧张素II受体(AT)拮抗剂(AT1受体拮抗剂)(氯沙坦)
5.5NO供体药物
临床常用NO供体药物主要有硝酸酯类和非硝酸酯类。
血管内皮舒张因子NO
5.6调节血脂药(HMG-CoA还原酶抑制剂:
洛伐他汀、氟伐他汀
苯氧乙酸类:
氯贝丁酯、非诺贝特、吉非罗齐
烟酸类:
烟酰胺)
8.6.1HMG-CoA还原酶抑制剂(他汀类)
他汀类药物抑制了该酶的活性,从实质上降低了内源性胆固醇的水平,
他汀类的活性形式:
酯键水解
HMG-CoA还原酶抑制剂的构效关系:
3,5-二羟戊酸(I部分)是抑制HMG-CoA还原酶的活性必须基团
5.6.2苯氧乙酸类
合成:
氯贝丁酯、吉非罗齐
苯氧乙酸类降血脂药的构效关系:
羧基为降脂活性必须基团,若为酯或酰胺衍生物活性不变,羧酸α碳上有两个甲基取代,或插入苯氧基可增强降脂活性。
中间连接链部分n的数目为0至3均有较好的降脂作用。
若氧原子被硫原子取代,可以增强降血脂活性。
吉非罗齐
第6章组胺受体拮抗剂及抗过敏和抗溃疡药
6.1组胺H1受体拮抗剂
经典型绝大部分有中枢副作用(镇静、防晕动、止吐)
分类:
乙二胺类(经典的H1受体拮抗剂,抗过敏药。
西尼二胺>曲吡那敏>芬苯
扎胺。
)、三环类(结构类似氯丙嗪,
镇静和安定副作用较明显,如异丙嗪)
西替利嗪作用特点:
引入亲水性羧甲氧烷基。
不易通过血脑屏障(BBB),镇静性大大减少。
盐酸苯海拉明作用特点:
中枢抑制作用显著。
还用于晕动及妊娠呕吐。
阿伐斯汀作用特点:
引入亲水的丙烯酸,,中枢副作用较小。
阿司咪唑作用特点:
非镇静H1受体拮抗剂(低BBB渗透率)
经典H1受体拮抗剂的构效关系(按上课记忆)
6.2组胺H2受体拮抗剂和抗溃疡药
抗溃疡药物配合抗微生物药物使用――幽门螺杆菌
6.3质子泵抑制剂
作用靶点:
H+/K+-ATP酶
第7章抗生素
分类:
1,β-内酰胺类;2,四环素类;3,氨基糖苷类;4,大环内酯类;5,氯霉素类
β-内酰胺类抗生素分为青霉素类、头孢菌素类、非经典的β-内酰胺类。
7.1β-内酰胺类
7.1.1青霉素类
稳定性:
β-内酰胺酶
和青霉素酰化酶
结构特征:
耐酸青霉素、
耐酶青霉素
广谱青霉素。
青霉素的构效关系
6位碳原子上的氢用甲氧基取代,可增加药物对β-内酰胺酶的抵抗能力。
2位羧基是活性必需基团,羧基为活性关键基团,去除和还原活性消失。
制成酯衍生物保留活性,且可增加口服吸收,长效化。
6位氨基的侧链可用不同的杂环进行取代,其活性可以得到增强。
当侧链的取代基含有极性基团,可扩大抗菌谱,增强其活性。
引入吸电子基团能提高药物对酸的稳定性,引入位阻基团能增加药物对β-内酰胺酶的抵抗能力。
7.1.2头孢菌素类
结构改造和基本结构:
7位酰胺侧链的取代基是抗菌谱的决定基团,可扩大抗菌谱并提高抗菌活性。
7位氢原子以甲氧基取代可增加β-内酰胺环的稳定性。
S原子可影响抗菌效力,将其改为碳或氧可提高抗菌活性。
3位取代基即可提高抗菌活性,又能影响药物代谢动力学的性质。
7.1.3β-内酰胺酶抑制剂
细菌产生的β-内酰胺酶使某些抗生素还未到达细菌作用部位即被酶水解失活,这是耐药的主要机制。
合成:
舒巴坦》
》
舒它西林(协同前药)(为改善舒巴坦的口服吸收,利用舒巴坦和氨苄西林碳2位
的羧基,用次甲基连成双酯,成为前药舒他西林,口服后
吸收迅速,在体内酯酶作用下水解为原药氨苄西林和舒巴
坦。
)
7.2四环素类抗生素
稳定性:
在酸性条件pH<2下,四环素类抗生素C-6上的羟基和C-5a上氢发生消除反应;在pH2-6下,C-4二甲胺基很易发生可逆的差向异构化
7.3大环内酯类抗生素
这类药物主要有红霉素
结构改造:
为增加红霉素对胃酸的稳定性,提高口服生物利用度,由于酸性条件下主要是C-9羰基和C-6羟基脱水环合,因此考虑将C-9羰基和C-6羟基进行保护。
罗红霉素是红霉素C-9肟衍生物,罗红霉素对胃酸有较好的稳定性
第8章合成抗菌药
分类:
磺胺类(磺胺嘧啶、磺胺甲恶唑)、喹诺酮类(诺氟沙星、左氟沙星、环丙沙星)
8.1、磺胺类和抗菌增效剂
设计思路:
wood–field抗代谢学说,又叫致死合成,主要目的是通过伪生物大分子干扰细菌或肿瘤细胞等的正常复制。
合成:
SMZ
磺胺类药物的构效关系:
对氨基苯磺酰胺为活性必需基团,邻、间位无效;
苯环若被其他芳环或芳杂环取代,或在苯环上引入其他基团,抑菌活性降低或丧失;
N4上一般无取代基,若有,则必须在体内易转化为游离氨基才有效,如RCONH-,R
N=N-,-NO2,等;
N1上为单取代,大多为吸电基,可使活性加强,如酰基、芳杂环。
N1,N1-双取代一般
丧失活性;
8.2、喹诺酮类抗菌药
喹诺酮类抗菌药的构效关系
3位COOH和4位C=O是活性必须基团,与DNA螺旋酶和拓扑异构酶Ⅳ结合,不可缺
少。
6位:
引入氟原子,与DNA旋转酶结合力增大,对细菌细胞壁的穿透力增大
7位取代活性增强(以取代或无取代的哌嗪、吡咯、吡咯烷基等五六原杂环较好
第9章抗肿瘤药
9.1烷化剂
9.1.1、氮芥类:
作用机制:
(1)生理pH7.4时,脂肪氮芥的β-氯原子离去生成乙撑亚胺离子,与DNA
的亲核中心起烷化作用,为双分子亲核取代反应(SN2)
(2)芳环与氮原子产生共轭作用,失去氯原子生成碳正
离子中间体,与DNA的亲核中心起烷化作用,为单分子亲核取代反应(SN1)
基本结构:
烷基化部分是抗肿瘤活性的功能基,载体部分改善药物在体内的吸收、分布和稳定性,提高选择性和抗肿瘤活性,降低毒性。
当R为脂肪烃时,推电子作用,为强烷化剂,选择性差
当R为芳烃时,拉电子作用,选择性增加,毒性降低。
合成:
环磷酰胺
卡莫司汀
9.2干扰DNA合成的抗肿瘤药-抗代谢类
分类:
嘧啶(设计思路:
(1)正常代谢产物是胞嘧啶核糖苷,利
用核糖的差向异构体阿拉伯糖,做成胞嘧啶拮抗物,当然实际还可修饰碱基(
(2)尿嘧啶掺入肿瘤组织的速度较其他嘧啶快,根据电子等排原理,以卤原子代替氢原子合成的卤代尿嘧啶衍生物中)
嘌呤
叶酸拮抗物
各类药物的设计思路
嘌呤拮抗物一般是模仿鸟嘌呤和腺嘌呤的代谢物次黄嘌呤
第10章药物设计(不考概念,若给出两个结构,要能够解释两个结构之间的联系,结构改造利用的是什么原理。
)
生物电子等排体
前药原理
电子等排体
孪药原理
插烯原理
先导化合物
 升级会员
升级会员 