基因工程试验.docx
《基因工程试验.docx》由会员分享,可在线阅读,更多相关《基因工程试验.docx(24页珍藏版)》请在冰豆网上搜索。
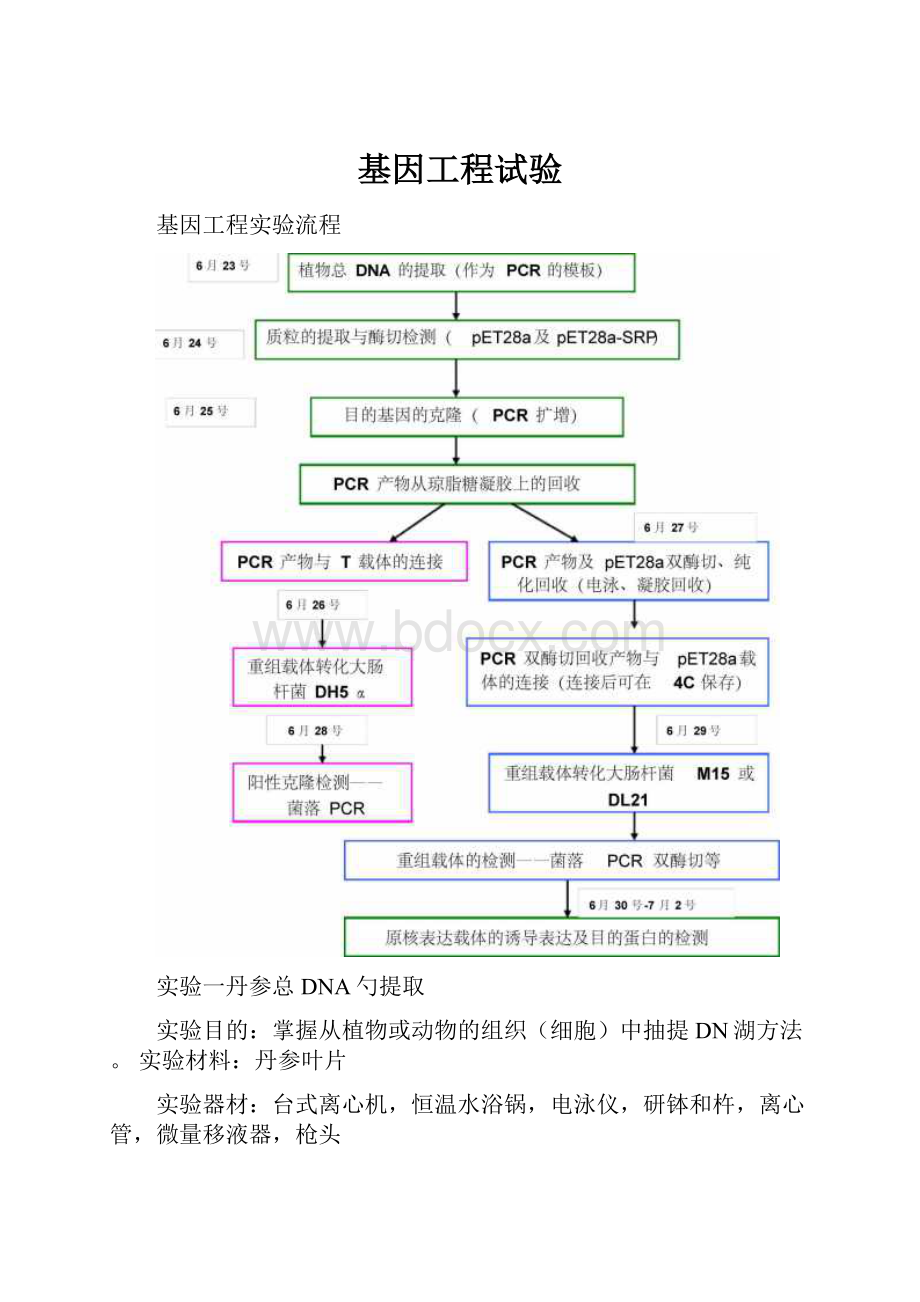
基因工程试验
基因工程实验流程
实验一丹参总DNA勺提取
实验目的:
掌握从植物或动物的组织(细胞)中抽提DN湖方法。
实验材料:
丹参叶片
实验器材:
台式离心机,恒温水浴锅,电泳仪,研钵和杵,离心管,微量移液器,枪头
实验步骤:
第一次使用前请先在漂洗液WEft结合液PQ中加入指定量的无水乙醇,充分混匀,加入后请及时在方框打钩标记已加入乙醇,以免多次加入!
取所需适量裂解液PL放置在65C预热,使用前加入6-筑基乙酿到终浓度2%
1.取适量植物组织(新鲜组织100mg或干重组织30m。
在研钵中加入液氮充分碾磨成细粉。
2.转移细粉到一个1.5ml离心管,不要解冻,加600vl65C预热的裂解液PL(确认已加入6-筑基乙醇至2%),剧烈涡旋振荡混匀,用大口径枪头轻柔吹打帮助裂解。
如果组织裂解困难,可根据需要加一个轻柔匀浆10秒的步骤帮助裂解。
3.65C水浴20-60分钟,在水浴过程中颠倒离心管以混合样品数次。
可选如果组织干燥或者产量低,可以适当延长水浴时间。
如RNA^留多,可在水浴前加入6^iIRNA酶(20mg/ml)。
4.加入700M氯仿/异戊酿(体积比24:
1混合),颠倒充分混
匀几分钟(或者涡旋混匀),13,000rpm离心5分钟。
若提取的
植物组织富含多糖多酚,可以在第4步前用等体积酚/氯仿
(1:
1)抽提一遍。
5.小心吸取上清到一个新的1.5ml离心管,注意不要吸到界面物质。
如上清比较浑浊,则需要重复步骤4一遍,直到得到透亮上清。
6.较精确估算上清量,加入1.5倍体积结合液PQ(请先检查是否已加入无水乙醇!
)后立刻涡旋,充分混匀。
此时可能出现沉淀,但不影响实验结果。
7.将上一步所得混合物(包括可能出现的沉淀)加入一个吸附柱AC中,(吸附柱放-5-入收集管中)13,000rpm离心30秒,倒掉收集管中的废液(先加700M离心,弃废液,再加入剩余的溶液,再次离心)。
8.加入500M抑制物去除液IR,12,000rpm离心30秒,弃废液。
9.加入700M漂洗液WB(请先检查是否已加入无水乙醇!
),12,000rpm离心30秒,弃掉废液。
10.加入500M漂洗液WB12,000rpm离心30秒,弃掉废液。
11.将吸附柱AC放回空收集管中,13,000rpm离心2分钟,尽量除去漂洗液,以免漂洗液中残留乙酿抑制下游反应。
12.取出吸附柱AC,放入一个干净的离心管中,在吸附膜的中间部位加100M洗脱缓冲液EB(洗脱缓冲液事先在65-70C水浴中预热),室温放置3-5分钟12,000rpm离心1分钟。
将得到的
溶液重新加入离心吸附柱中,室温放置2分钟,12,000rpm离心
1分钟。
洗脱体积越大,洗脱效率越高,如果预计和需要产量高,可增大洗脱体积,如果需要DNA^度较高,可以适当减少洗脱体积,但是最小体积不应少于50vl,体积过小降低DN碱脱效率,减少DNA^量。
13.DNA可以存放在2-8C,如果要长时间存放,可以放置在一20°C。
按照植物总DNA提取试剂盒操作说明。
1%琼脂糖凝胶电泳检测
注意事项:
1.所有的离心步骤均在室温完成,使用转速可以达到13,000rpm的传统台式离心机,如Eppendorf5415C或者类似离心机。
2.开始实验前将需要的水浴先预热到65C备用。
3.需要自备氯仿/异戊酿(体积比24:
1混合)、无水乙醇和6
-筑基乙醇。
4.结合液PQ和抑制物去除液IR中含有刺激性化合物,操作时要戴乳胶手套,避免沾染皮肤、眼睛和衣服。
若沾染皮肤、眼睛时,要用大量清水或者生理盐水冲洗。
5.不同来源的植物组织材料中提取DNA的量会有差异,一般100mg新鲜组织典型产量可达3-25vgo
6.洗脱液EB不含有螯台剂EDTA不影响下游酶切、连接等反应。
也可以使用水洗脱,但应该确保pH大于7.5,pH过低影响洗脱
效率。
用水洗脱DNA^该保存在—20Co
实验结果:
本组实验结果是第九孔和第十孔,其中第九孔的DNA
提取效果较第十孔提取效果好。
两空之间的差异可能是由于两个
离心管中添加的植物量不一样,所以最后提取的量不一样。
实验结果分析:
1.在液氮研磨叶片的时候,研磨不够充分,且在向离心管中添加
的时候,添加的量不多,速度慢,导致氧化,对提取效率造成很大影响。
2.可能是因为在提取DNA的最后步骤向吸附管中添加的洗脱液太多150ul,导致DNA的浓度过低,所以条带不亮。
3.在第九步中,将吸附柱放置室温晾干,可能晾置时间不足,导
致添加的有机溶剂没有完全挥发,而有机溶剂影响了DNA的提
取。
实验二质粒DNA的提取及酶切检测
实验目的:
学习和掌握小量提取质粒的方法。
实验原理:
质粒提取(碱裂解法)根据共价闭台环状质粒DNA
与线性染色体DNA在拓扑学上的差异来分离它们。
在pH12.0-12.6的碱性条件下,线性的染色体DNA变性,双螺旋解开,而共价闭环质粒DNA仍保持双链紧密缠绕的状态。
将pH
调至中性并保持高盐浓度的条件下,染色体DNA之间形成不落
性网状结构,大部分DNA和蛋白质在去污剂SDS作用下形成沉淀,而共价闭环质粒DNA保持可洛状态,经离心可将大部分细胞碎片、染色体DNA、RNA及蛋白质除去,质粒DNA留在上清液中,用酚、氯仿抽提,可进一步纯化质粒DNAo
实验材料:
含有pET28a质粒的大肠杆菌H5a;构建好的含有目的基因SRP的pET28a-SRP质粒的大肠杆菌DH5a实验器材:
离心机、水浴锅、电泳仪、离心管,微量移液器,枪头,等
实验步骤:
质粒提取按照试剂盒操作说明。
1.用1.5ml离心管收集1-5ml菌液。
12,000rpm离心1min,弃上清。
应根据所培养菌体的浓度与质粒的拷贝数,确定收集的菌液量。
菌量过大可能导致洛菌不充分,纯化时会影响质粒纯度。
菌
体的体积与质粒拷贝数参见注意事项。
2.250vl溶液I/RNaseA混合液,漩涡剧烈振荡直至菌体完全重新悬浮。
室温静置1-2min。
初次使用本试剂盒时,请将
RNaseA全部加入到溶液I中,均匀混合后于4C保存。
可保存6个月。
•不要残留细小菌块。
菌体悬浮充分与否将决定质粒DNA得率
的高低。
•室温静置1-2min是为使溶液中的RNA被充分降解。
3.加入250vl溶液II,轻柔地反复颠倒混匀6-10次。
室温放置1-2min,使菌体充分裂解,直至形成澄清的裂解溶液。
•若溶液H出现沉淀,请于37C保温溶解。
待恢复至室温后使
用。
沉淀的出现不会影响质粒DNA的纯化结果。
•不可剧烈混和,否则会使染色体DNA断裂。
•此步骤不宜超过5min。
4.加入350vl溶液山,立即轻柔地反复颠倒混匀5-6次。
此时会出现白色絮状沉淀。
5.12,000rpm室温离心10min,收集上清。
6.将上清置于DNA纯化柱中,静置1-2min。
•如果收集的上清液过多,超过DNA纯化柱容积(800vl),可将上清分次加入DNA纯化柱中。
7.12,000rpm离心1min,弃滤液。
•此时质粒DNA被吸附于DNA纯化柱的硅胶膜上。
8.加入500M溶液PB/PE(去蛋白液),12,000rpm离心1min,弃滤液。
•此步骤的作用是将硅胶膜上吸附的蛋白、盐等杂质洗脱,以获
得高质量质粒DNAo
9.加入500M溶液WB,12,000rpm离心1min,弃滤液。
•溶液W初次使用前用无水乙醇按1:
1.5稀释,即含60%乙醇。
10.加入500M溶液w,12,000rpm离心1min,弃滤液。
11.12,000rpm离心3min,以彻底去除纯化柱中残留的液体。
12.将DNA纯化柱置于新的离心管中。
向纯化柱中央处,悬空滴加50-100M溶液Eluent,室温放置2min。
•12,000rpm离心1min,管底即为高纯度质粒DNA。
质粒DNA
于-20C保存
•溶液Eluent可用无菌双蒸水代替,但其pH需为8.0-8.5。
溶液Eluent的加入体积视质粒拷贝数多少、用户对质粒浓度要求而
o
酶切反应体系:
载体pET28a-SRP30ul
XhoI1ul
HindIII1ul
双酶切buffer5ul
超纯H2O13ul
37度,3小时。
电泳检测分析
注意事项;
1.使用前往准备好的solution中加入RNaseA;
2.浓缩的washbuffer用乙酿稀释;
3.所有步骤都在室温下操作。
实验结果:
实验结果分析
实验三目的基因的克隆
[实验目的]
通过本实验学习PCR反应的基本原理与实验技术。
[实验原理]
多聚酶链式反应(polymerasechainreaction,PCR)的原理类似于DNA的天然复制过程。
在待扩增的DNA片段两侧和与其两侧互补的两个寡核昔酸引物,经变性、退火和延伸若干个循环后,DNA扩增2n倍。
1,变性:
加热使模板DNA在高温下(94C)变性,双链间的氢键断裂而形成两条单链,即变性阶段。
2.退火:
使溶液温度降至50-60C,模板DNA与引物按碱基配对原则互补结合,即退火阶段。
3.延伸:
溶液反应温度升至72C,耐热DNA聚合酶以单链DNA为模板,在引物的引导下,利用反应混合物中的4种脱氧核昔三磷酸(dNTP),按5,+3,方向复制出互补DNA,即引物的延伸阶段。
上述3步为一个循环,即高温变性、低温退火、中温延伸3个阶段。
从理论上讲,每经过一个循环,样本中的DNA量应该
增加一倍,新形成的链又可成为新一轮循环的模板,经过25-30个循环后DNA可扩增106-109倍。
典型的PCR反应体系由如下组分组成:
DNA模板、反应缓
冲液、dNTP、MgCl2、两个合成的DNA弓I物、耐热Tag聚合酶。
[仪器、材料与试剂]
(一)仪器
1.PCR热循环仪2.琼脂糖凝胶电泳系统3.吸
头、0.2ulPCR管,微量移液器,
(二)材料:
质粒
(三)试剂
1.2XPCRMasterMix(含dNTP,Tag酶,缓冲液)
2.质粒DNA
3.弓I物(上、下游引物浓度均为约10umol/ul)
F:
5-CCCAAGCTTCTTTTTAATCCTTCCCTCAAATATC-3(HindIII)
R:
5-CCGCTCGAGGGGACAATGAAAAAGCTGATAAC-3(XhoI)
4.灭过菌的millipore水
5.琼脂糖
6.DNA相对分子质量标准物DL2000
[实验方法]
1.学习PCR仪的使用,设定反应程序:
反应程序为:
94C变性3min;94C变性30s,57C退火30
s,72°C延伸1min20s,30个循环;72C延伸8min。
4C保存
2.加样(冰上操作):
50ul反应体系,其中上游引物1ul,下游引物1ul,丹参DNA
模板2ul或质粒DNA模板1ul,2XPCRMasterMix25ul,灭菌水
21或22ul
3.瞬时离心后在PCR仪上进行扩增反应。
4.电泳检测。
注意事项:
1.在PCR仪设定的时候要注意时间与温度,不要设置错误;
2.在加样的时候要确保加入的量准确,在冰上加样;
3.操作时设立阴阳性对照和空白对照,即可验证PCR反应的可
靠性,又可以协助判断扩增系统的可信性;
4.PCR之后最好能立刻电泳,不要隔夜。
实验四PCR产物从琼脂糖凝胶上的回收
仪器:
水浴锅,离心机,电子天平,微量移液器
试剂:
琼脂糖凝胶回收试剂盒
耗材:
Eppendorf管,大小枪头
实验步骤:
按照DNA®脂糖凝胶回收提取试剂盒操作说明。
1.在长波紫外灯下,用干净刀片将所需回收的DN燎带切下,
尽量切除不含DNA勺凝胶,得到凝胶体积越小越好。
2.将切下含有DN成带凝胶放如1.5ml离心管,称重。
先称一个空1.5ml离心管重量,然后放入凝胶块后再称一次,两次重量相减,得到凝胶的重量。
3.加一到二倍体积溶胶/结合液DR如果凝胶重为0.1g,其体积可视为100则加入100~200M溶胶液。
凝胶块最大不能超过400mg如果超过400mg请用多个离心柱,进行回收。
4.56°C水浴放置5min(或直至胶完全溶解)。
每1-2min涡旋震荡一次帮助加速洛解。
5.将上一步所得溶液加入吸附柱AC中(吸附柱放入收集管
中),12000rpm离心30-60S,倒掉收集管中的废液。
如果总体
积超过750M,可分两次将溶液加入同一个吸附柱AC中。
6.加入700vl漂洗液WB12000rpnW心min,弃掉废液。
7.将吸附柱AC放回空收集管中,12000rpm离心2min,尽量除去漂洗液,以免漂洗液中残留乙酿抑制下游反应。
8.取出吸附柱AC,放入一个干净的离心管中。
在吸附膜的中
间部位加50M洗脱缓冲液EB(洗脱缓冲液事先在65-70C水浴中加热效果更好),室温放置2min,12000rpm离心1min。
如果需要较多量DNA可将得到的溶液重新加入离心吸附柱中,
离心1min。
洗脱体积越大,洗脱效率越高,如果需要DN缺度较高,可以适当减少洗脱体积,但是最小体积不应少于30vl,体积过小降低了DNA洗脱效率,减少产量。
回收的PC产物进行琼脂糖凝胶电泳检测,—20C保存或直接用于后续试验。
注意事项:
1:
保证是新胶新缓冲液浓度大小合适
2:
割胶大小要对,不要割错了条带,这很重要,
3:
短波紫外线(254)会引起DNA的断裂或是形成TT二聚体,前者会使以后的链接与转化等实验失败,后者肯能造成基
因突变,给克隆工作带来麻烦,波紫外线,回收DNA时也要尽量短时照射,避免连续长时间照射
4:
回收时要尽量的柔缓,避免将DNA链弄断
5:
做胶时要尽量小薄,回收时割胶尽量少提高回收效率乙酿对回收的影响较大应注意吸干。
实验结果:
本组为第五孔
实验结果分析:
本组为第五孔。
DNA的提取量较大,且PCR较成功。
电泳中条带很亮很清晰,说明目的DNA已经被大量克隆,可以进行下一步PCR产物与T载体的连接。
PCR产物从琼脂糖凝胶上的回收这一步骤比较成功,主要
因为:
1.PCR的结果比较好,获得了大量的目的基因;
2.跑胶的时间以及胶自身制备好;
3.在切胶的时候,切得大小比较合适,所以提取的回收率较高。
实验五PCR产物与T载体的连接
[实验目的]
将通过PCR获得的目的基因与载体进行连接,可进一步用于测序和克隆等。
[实验原理]
所用的T载体为线装载体,其两端分别有突出的一个T;而
1926
X-nl:
XF.-
I'P,MI14鹏
BsalU77Gsu114969pmI1495
PCR产物在其两端各有一个A末端,因此,可将目的基因与载体连接起来。
而且T载体中含有LacZ基因,如果有外源基因的插入,则LacZ基因被破坏,便于通过蓝白斑进行后面的筛选。
/PstlB4
..fCa90
C*,Sal190多”.NdelK
Ms-1123
RsaII朋弟Sealim
.BxtXI1tl
[实验试剂]
pGEM-T载体、T4DNA连接酶、T4DNA连接酶缓冲液、
回收的PCR产物
[实验方法]
1.将pGEM-T载体于离心机上进行短暂的离心,使载体都
位于管低;
2.于Eppendorf管中加入5ulT4DNA连接酶缓冲液、1ulpGEM-T载体、1ulT4DNA连接酶、3ul回收的PCR产物;
3.将所加的物质于离心机上稍加离心,充分混匀,室温保育
3小时或4°C过夜(效果更好)。
注意事项:
1.使用之前,将pGEM-T载体于离心机上进行短暂的离心。
2.所有的样品再添加的时候都应该在冰上操作,保持样品的活性。
加量的时候要准确。
3.将所有的物质充分混匀,使其充分接触,提高连接效率。
4.放在PCR仪中过夜,使其充分连接。
实验六重组载体转化大肠杆菌
[实验目的]
将质粒DNA转化到大肠杆菌中。
[实验试剂]
LB培养基(固体和液体)Amp(10mg/mL)
X-Gal(20mg/ml)IPTG(0.2mg/ml)感受态细胞DH5a
[实验器材]
摇床,离心机、水浴锅
[实验步骤]
1.取感受态细胞DH5a(100uL)用时在冰上缓慢解冻10min后立即使用。
2.每管中加入PCR与T载体的连接产物(10uL)。
于冰上保温40min。
3.于42C热激80s,中间保持静止。
4.迅速在冰上冷却2min,而后加入400-500ulLB液体培养基,37C100—120rpm振荡培养40-60min。
5.将细菌铺布于含IPTG/X-Gal(4ul/40ul)的LB固体培养基(含Amp50ug/ml)上,在37C培养过夜。
于24小时内挑取白色单菌落,在含Amp50ug/ml的LB液体培养基中过夜培养。
注意事项:
1.制备感受态细胞的全部操作均须至于冰浴操作,同时注意近
火无菌操作,防止感受态细胞受杂菌污染;
2.42C热处理时很关键,转移速度要快且温度要准确;
3.菌液涂皿操作时,避免反复来回涂步,因为感受态细胞壁发
生了变化,过多的机械挤压会使细胞破裂,影响转化率。
实验结果:
LB固体培养基(含Amp50ug/ml)上,在37C培养过夜之后没有长出单菌落,实验失败。
实验结果分析:
单菌落没长出来,原因可能有
(1)PCR产物与T载体的连接实验没有做好,产物没有很好与
载体连接,导致重组载体转化大肠杆菌失败;
(2)感受态细胞需要在冰上加样,可能在实验过程中将感受态
细胞取出,使其感受态受到影响;
(3)实验在混匀的时候使用漩涡混匀器,可能对细胞有破坏。
实验七阳性克隆检测——菌落PCR
[实验目的]将通过PCR检测重组大肠杆菌的正确性。
[实验原理]菌落PCR与我们通常的普通的PCR的不同在于,不用提取基因组DNA,而是直接以菌体热解后暴露的DNA
直接为模板进行PCR扩增,是一种可以快速鉴定菌落是否为含目的质粒的实验方法。
该方法操作简单,快捷,阳性率较高,常用于在转化鉴定中。
[实验试剂]
1.2XPCRMasterMix(含dNTP,Tag酶,缓冲液)
2.菌液
3.弓I物(引物溶液浓度10pmol/L)
F:
5-CCCAAGCTTCTTTTTAATCCTTCCCTCAAATATC-3(HindIII)
R:
5-CCGCTCGAGGGGACAATGAAAAAGCTGATAAC-3(XhoI)
4.灭过菌的millipore水
5.琼脂糖
6.DNA相对分子质量标准物DL2000
[实验操作]
1.取菌液20ul,100C水浴10min(或在PCR仪(反应程序为:
100°C变性10min;4C保存))。
2.PCR体系配制加样(冰上操作)
20ul反应体系,其中上游引物0.5ul,下游引物0.5ul,煮沸后菌液2ul,2XPCRMasterMix10ul,灭过菌的millipore水7ulo
3.设定PCR反应程序:
反应程序为:
94C变性3min;
94C变性30s,57C退火30s,72C延伸1min20s,30个循环;
72°C延伸8min。
4C保存
4.瞬时离心后在PCR仪上进行扩增反应。
5.电泳检测。
实验八原核表达载体pET28a-SRP的构建
实验目的:
学习和掌握原核表达载体的构建方法与流程。
实验原理:
通过目的基因及表达载体的酶切、酶连反应,构建重组表达载体pET28a-SRP并将其导入大肠杆菌M15菌株。
实验材料:
重组载体pGEM-T-SRP,目的基因PCR克隆的凝胶回收产物,载体pET28a。
实验步骤:
1.双酶切(50ul反应体系)
(1)PCR凝胶回收产物30ul,XhoI1ul,HindIII1ul,buffer
5ul,H2O13ul
(2)载体pET28a30ul,XhoI1ul,HindIII1ul,buffer5ul,
H2O13ul
37度,反应3小时。
2.电泳检测,凝胶回收(按照凝胶回收试剂盒操作说明)
注:
双酶切体系
(1)回收约1000bp的目的基因,
(2)将双酶切的pET28a回收。
3.连接反应
于Eppendorf管中加入1ulT4DNA连接酶缓冲液、1ulT4
DNA连接酶、5ul酶切后回收的pET28a载体、3ul回收的目的基因,瞬时离心。
16度,3小时或4度,过夜。
4.连接产物转化大肠杆菌M15(方法参照实验六)
17prareitM~
T7traiscrijrtifflistart
HiE*Ts£ccdi尊sequence27D-28TI
TT*Tag也
MulliplaelwiresitesgIU血E156H2Q3
Hu-Tig:
codirijrequench140-L5?
T7tenunwtor2ME
亨卯切cwT7>t®52
arifin32跪
Xanwdipcnq,ucncc399&-4301
flori^n4»0>53S8
XhOg]NQtht«>EAgl^WiFfie通1问SelipTBlSael(WiEeci丽顷EiAmHlnos-
ThefarpET-28b(+)andj£T-2ScWar«1htojhvkpEP23a(+)(stnnn)withth±±S£ceptiOn£;^ET-26b(+)if加
S节嘛plasnd:
subtractLbpfron>tach由甲beyord瓦鱼HIst】瓯j£T-20c(+)iis&536TbpplasKtd:
■suhtrF2bpfmeadtsitebeytoldl岛sHIatI9&
Bpu11D2l:
«
E«i£7ir-<
-.1^11
I4wkZJ1lrwe>为印Neo
EWLU111MEapIg白esiiozi»5immi:
3hw
0$il:
'4-FSpl:
携.
MJJ
UU
■■'edi
〔H
pET-2fla(*)
(536Sbp!
BssSIsas-
tS0hl£BB.
IjJ
■':
f0«HHW
」EcdR
>Awpaii(&
实验九原核表达载体的诱导表达及目的蛋白的检测
目的:
学习原核表达的诱导原理和方法。
原理:
将外源基因克隆在含有lac启动子的pET28a表达载体中,让其在E.coli中表达。
先让宿主菌生长,lacI产生的阻遏蛋白与lacI操纵基因结合,从而不能进行外源基因的转录与
 升级会员
升级会员 