心脏的电生理学基础Word文档格式.docx
《心脏的电生理学基础Word文档格式.docx》由会员分享,可在线阅读,更多相关《心脏的电生理学基础Word文档格式.docx(13页珍藏版)》请在冰豆网上搜索。
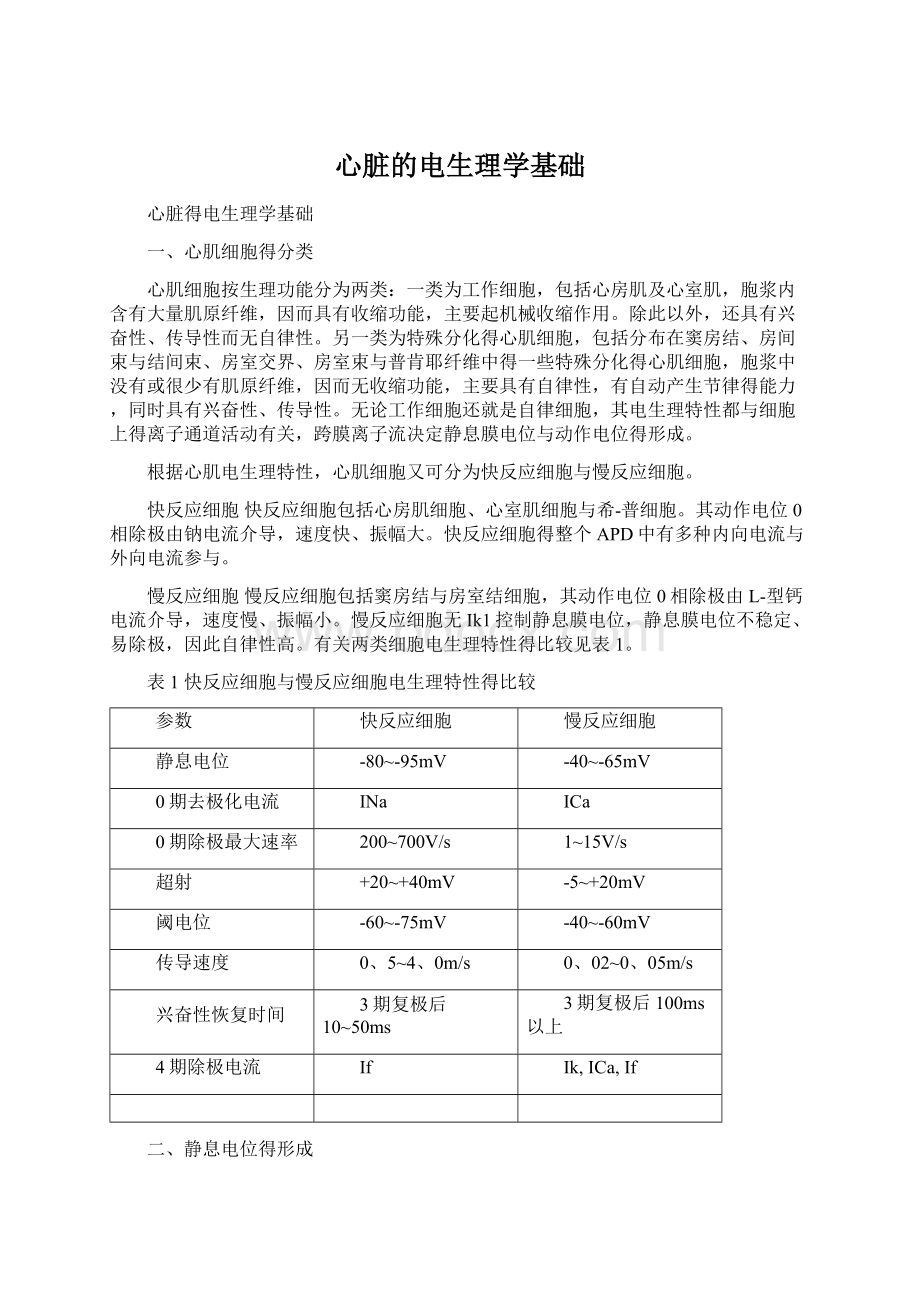
1~15V/s
超射
+20~+40mV
-5~+20mV
阈电位
-60~-75mV
-40~-60mV
传导速度
0、5~4、0m/s
0、02~0、05m/s
兴奋性恢复时间
3期复极后10~50ms
3期复极后100ms以上
4期除极电流
If
Ik,ICa,If
二、静息电位得形成
静息电位(restingpotential,RP)就是指安静状态下肌细胞膜两侧得电位差,一般就是外正内负。
利用微电极测量膜电位得实验,细胞外得电极就是接地得,因此RP就是指膜内相对于零得电位值。
在心脏,不同组织部位得RP就是不相同得,心室肌、心房肌约为-80~-90mV,窦房结细胞-50~-60mV,普肯耶细胞-90~-95mV。
各种离子在细胞内外得浓度有很大差异,这种浓度差得维持主要就是依靠位于细胞膜与横管膜上得离子泵。
如Na-K泵(Na-Kpump),也称Na-K-ATP酶,其作用将胞内得Na+转运至胞外,同时将胞外得K+转运至胞内,形成细胞内外Na+与K+浓度梯度。
Na-K-ATP酶得磷酸化需要分解ATP,通常每分解一分子ATP可将3个Na+转运至膜外,同时将2个K+转运至膜内。
心肌细胞外Ca2+([Ca2+]0)与细胞内Ca2+([Ca2+]i)相差万倍,维持Ca2+跨膜浓度梯度得转运系统其一就是位于细胞膜上得Na+/Ca2+交换体(Na+/Ca2+exchanger),它得活动可被ATP促进,但不分解ATP,因而也不直接耗能。
Na+/Ca2+交换体对Na+与Ca2+得转运就是双向得,可将Na+转入胞内同时将Ca2+排出胞外(正向转运),也可将Na+排出而将Ca2+转运至胞内(反向转运)。
转运得方向取决于膜内外Na+、Ca2+浓度与膜电位。
无论就是正向还就是反向转运,其化学计量学都就是3个Na+与1个Ca2+得交换,Na+/Ca2+交换电流(INa/ICa)为内向电流,电流方向与Na+流动得方向相一致,Na+内流而Ca2+外排。
经Na+/Ca2+交换排出Ca2+得过程就是间接地以Na泵得耗能活动为动力得。
另一个维持Ca2+跨膜梯度得转运系统就是位于肌质网(sarcoplasmicreticulum,SR)膜上得Ca泵起着主要作用。
Ca泵也称Ca-ATP酶,它每分解一分子ATP可将胞浆中2个Ca2+逆电化学梯度转动至SR内,使[Ca2+]i降低到0、1µ
mol·
L-1以下。
心肌细胞膜上也存在Ca-ATP酶,可逆电化学梯度将胞浆内Ca2+转运至胞外。
带电功率离子得跨膜流动将产生膜电位得变化,变化得性质与幅度决定于电流得方向与强度。
离子电流得方向就是以正电荷移动得方向来确定得;
正电荷由胞外流入胞内得电流为内向电流,它引起膜得去极化;
正电荷由胞内流出胞外得电流称为外向电流,它引起膜得复极化或超极化。
心室肌、心房肌得RP能保持稳定,就是由于静息状态下内向电流与外向电流大小相等,电荷在膜两侧得净移动为零。
决定RP得离子电流主要就是Na+与K+。
原因就是静息状态下膜对Ca2+几乎没有通透性,其作用可以忽略。
Cl-就是一个被动分布得离子,它不决定RP,而就是RP决定它得分布。
以上分析表明一个稳定得RP,其外向得K+电流与内向得Na+电流相等。
RP主要取决于膜得K+电导与Na+电导。
膜对哪一种离子得电导更大,RP就更接近哪一种离子得平衡电位。
静息时,K+电导》Na+电导,RP接近于K+平衡电位。
三、心肌细胞动作电位得产生机制
动作电位(actionpotential,AP)就是指一个阈上刺激作用于心肌组织可引起一个扩布性得去极化膜电位波动。
AP产生得基本原理就是心肌组织受到刺激时会引起特定离子通道得开放及带电离子得跨膜运动,从而引起膜电位得波动。
由于不同心肌细胞具有不同种类与特性得离子通道,因而不同部位得心肌AP得开关及其它电生理特征不尽相同。
(一)心室肌、心房肌与普肯耶细胞动作电位
心室肌、心房肌与普肯耶细胞均属于快反应细胞,AP形态相似。
心室肌AP复极时间较长(100~300ms),其特征就是存在2期平台。
AP分为0,1,2,3,4期。
0期:
除极期,膜电位由-80~-90mV在1~2ms内去极化到+40mV,最大去极化速度可达200~400V/s。
产生机制就是电压门控性钠通道激活,Na+内流产生去极化。
1期:
快速复极早期,膜电位迅速恢复到+10±
10mV。
复极得机制就是钠通道得失活与瞬间外向钾通道Ito得激活,K+外流。
在心外膜下心肌Ito电流很明显,使AP出现明显得尖锋;
在心内膜下心肌该电流很弱,1期几乎瞧不到。
2期:
平台期,形成得机制就是内向电流与外向电流平衡得结果。
平台期得内向电流有ICa-L,INa+/Ca2+,以及慢钠通道电流。
其中最重要得就是ICa-L,它失活缓慢,在整个平台期持续存在。
INa+/Ca2+在平台期就是内向电流,参与平台期得维持并增加平台得高度。
慢钠通道电流就是一个对TTX高度敏感得钠电流,参与平台期得维持。
参与平台期得外向电流有Ik1,Ik与平台钾通道电流Ikp。
ICa-L得失活与Ik得逐渐增强最终终止了平台期而进入快速复极末期(3期)。
3期:
快速复极末期,参与复极3期得电流有Ik,Ik1与生电性Na泵电流。
3期复极得早期主要就是Ik得作用,而在后期Ik1得作用逐渐增强。
这就是因为膜得复极使Ik1通道开放得概率增大,后者使K+外流增加并加速复极,形成正反馈,使复极迅速完成。
4期:
自动除极期(又称舒张期自动除极期),主要存在于自律细胞,如普肯耶细胞与窦房结细胞。
普肯耶细胞4期除极得最重要得内向电流为If电流。
由于它激活速度较慢,故它得4期除极速率较慢。
在普肯耶细胞4期除极得后期,稳态得Na+窗电流参与自动除极过程。
窦房结细胞参与4期除极得离子有延迟整流钾电流(Ik),起搏电流(If),电压门控性ICa-L,ICa-T。
这些离子电流没有一个能独立完成窦房结得4期除极,外向Ik衰减,相当于内向电流逐渐加强,在4期除极中起主要作用,也就是4期除极得主要机制;
If超极化激活,故在膜电位负值较大得细胞起较大作用;
Ca2+内流主要参与4期后半部分得除极。
心房肌动作电位与心室肌相比,主要特点就是:
①1期复极较迅速,平台期不明显,因为心房肌Ito电流较强而ICa-L较弱;
②3期复极与静息期有乙酰胆碱激活得钾通道KAch参与。
普肯耶细胞属于快反应自律细胞,其AP与心室肌相比一个显著区别就是具有4期自动除极过程。
普肯耶细胞Ik1电流较强,RP可达-90mV。
0期最大除极速率高;
它得Ito电流较强,1期复极速度较快;
它得平台期持续时间长,可达300~500ms。
(二)窦房结与房室结细胞动作电位
窦房结细胞属于慢反应细胞,其AP与心室肌相比一个特点就是0期去极化幅度小,没有1期与2期,由0期直接过渡到3期,也具有4期自动除极过程。
另一个特点就是窦房结产生AP各时相得离子电流也与快反应细胞不同。
0期去极化就是ICa-L激活引起得,激活过程较慢,故0期得去极化速度低。
3期复极主要就是由于ICa-L得失活与Ik得激活形成得,IKAch也参与了3期复极。
房室结细胞AP得0期除极速度与幅度略高于窦房结,而4期去极化速度较低。
四、心肌细胞得电生理特性
(一)兴奋性
1.心肌兴奋性得产生机制
兴奋性(excitability)就是指心肌细胞受刺激后产生动作电位得能力。
包括静息电位去极化到阈电位水平以及有关离子通道得激活两个环节。
对快反应细胞来说,形成AP得关键就是钠通道得激活。
当静息电位绝对值高于80mV时,所有钠通道都处于可开放状态,接受阈刺激即可产生动作电位。
随着膜得去极化,电压门控钠通道开放得概率增大,当刺激能使膜电位去极化到某一临界值时,这一临界值称为阈电位(thresholdpotential),内向钠电流得强度充分超过了背景外向电流使膜迅速去极化形成AP得0期。
慢反应细胞形成AP得关键就是钙通道得激活而产生得。
2.影响兴奋性得因素
心肌兴奋性主要取决于静息膜电位得大小及阈电位水平。
静息膜电位绝对值减小,阈电位水平下降均能提高心肌兴奋性。
其中阈电位水平就是最重要得。
决定阈电位得主要因素就是钠通道得机能状态。
虽然钠通道得关闭状态与失活状态都就是不导通得,但它们对兴奋性得影响却就是截然相反得。
关闭状态得通道越多,兴奋性越高;
而失活状态通道所占得比例越大,细胞就越不容易兴奋。
在此处简述一下钠通道得三种机能状态。
根据钠通道得Hodgkin-Huxley(H-H)工作模型,电压依赖性钠通道受膜电位得影响,在不同电压影响下,通道蛋白发生构象变化而使通道不断转换于静息态(restingstate)、开放状态(openstate)与失活状态(inactivestate)。
通道内侧有m激活闸门与h失活闸门来控制通道得开启与关闭(图6-1-2)。
静息时,m门位于通道内,使通道处于关闭状态,即静息态;
兴奋时,在去极化作用下,m闸门激活而移出通道外,使通道开放,Na+内流,即为激活态;
但在去极化作用下,原来位于通道外得h闸门也被激活,而以稍慢得速度移到通道内部,从而使通道开放瞬间后失活而关闭,即为失活态;
随后在膜电位复极化得作用下,m与h闸门又逐渐移到原来得位置,即m闸门位于通道内,h闸门位于通道外,进入静息状态,此时兴奋恢复正常。
单从电压依赖性上瞧,两个闸门几乎没有同时开放得可能性,但两个闸门得动力学参数相关很大,激活门开放得时间常数τm比失活门关闭得时间常数τh小得多,若刺激使膜从静息状态迅速去极化时,激活门迅速开放而失活门还未来得及关闭,钠通道便进入两个闸门都开放得激活状态,此时Na+内流。
随着失活门随后得关闭,钠通道便进入失活状态。
失活关闭状态得通道不能直接进入开放状态而处于一种不应期。
只有在经过一个额外刺激使通道从失活关闭状态进入到静息关闭状态后,通道才能再度接受外界刺激而激活开放。
这一过程称为复活(recovery)。
钠通道得膜电位在-80~-90mV时,几乎全部通道都处于关闭状态,一旦迅速去极化,钠通道开放得概率也很高,较低程度得去极化就可以激活钠通道,因而阈电位较低(负值较大),兴奋性较高。
随着静息电位得减小,失活闸门逐渐关闭或进入失活状态得钠通道越来越多,需较强得去极化才能激活钠通道,阈电位上移,兴奋性逐渐降低甚至消失。
即RP得减小超过一定程度时阈电位会上移,使RP与阈电位得差距增大,兴奋性减小甚至消失。
高血钾对心肌兴奋性得影响就就是一个典型得实例。
轻度高血钾使RP略微减小(如从-90mV减少至-80mV)时,阈电位无显著变化,RP与阈电位差距减少,故兴奋性升高;
重度高血钾时RP进一步减小而使阈电位升高,兴奋性则降低。
此外,某些因素(如药物)通过改变钠通道激活与失活过程而影响兴奋性。
例如1类抗心律失常药可使钠通道稳态失活曲线左移,阈电位上移,兴奋性降低。
3.兴奋性得恢复
心肌兴奋后,兴奋性暂时丧失,随着复极过程得进行,兴奋性又逐渐恢复,其机制为随着膜电位得增大,失活状态得钠通道或钙通道逐步进入关闭状态,即复活过程。
复活就是电压与时间依赖性得,在快反应细胞,钠通道复活过程为电压依赖性,根据复极过程中膜电位得变化,将心肌复极过程中得兴奋性分为以下几期:
①绝对不应期,终止于3期复极至-55mV左右,此期钠通道全部处于失活状态,不产生兴奋。
②有效不应期,从0期开始终止于3期-66mV左右,比绝对不应期稍长,在此期得后段,强刺激可引起局部兴奋,但不产生扩布性得AP。
③相对不应期,3期复极从-60mV至-80mV期间,此期有部分钠通道复活,兴奋性逐渐恢复,较强刺激有可能引起AP。
④超常期,相当于3期复极至-80mV~-90mV之间,此期钠通道已近乎全部复活。
在慢反应细胞,兴奋性得恢复表现为较大得时间依赖性,兴奋性得恢复滞后于膜电位得恢复。
(二)自律性
自律性(automaticity)就是指细胞在没有外界刺激得条件下自动地产生节律性兴奋得特性。
通常以单位时间内产生AP得次数来衡量自律性得高低。
自律性产生得机制就是4期自动除极,参与4期自动除极得离子流前已叙述,最终结果形成一个净内向电流而使膜去极化。
在正常心脏,窦房结得自律性最高,70~80次/min;
其次就是房室交界,40~60次/min;
心室传导系统自律性最低,15~40次/min。
由于窦房结自律性最高,每当其它自律组织得兴奋还没有发放之前,窦房结得冲动已经扩布下来,而兴奋后得心肌细胞暂时处于不应期状态,导致其它自律组织得起搏活性始终表现不出来,成为潜在起搏点。
窦房结为心脏得正常起搏点(pacemaker)。
当窦房结病变,自律性降低到潜在起搏点之下,或就是它所发放得冲动不能下传时(如窦房阻滞、房室传导阻滞),潜在起搏点有可能成为有效起搏点而发放冲动,形成异位心律(室性心律、交界性心律等)。
潜在起搏点得自律性升高超过窦房结,将出现快速性心律失常。
(三)传导性
传导性(conductivity)心肌细胞膜得任何部位产生得兴奋不但可以沿整个细胞膜扩布,且可通过细胞间缝隙连接(gapjunction)传导到另一个心肌细胞,从而引起整个心脏得兴奋与收缩。
窦房结发出得兴奋首先经心房肌与心房肌中得几条细小得传导束(房间束与结间束)传向房室与整个心房,再经房室交界到达房室束。
兴奋进入心室传导系统后,沿走行于心内膜下得左束支与右束支及其进一步分支形成得普肯耶纤维,传导至心内膜下心肌,再传至心外膜侧。
兴奋由窦房结发出经上述途径传遍整个心脏,总共约需时0、22s。
心脏传导性由0期去极化速度与幅度决定。
快反应细胞0期除极化速率由钠内流决定,慢反应细胞0期除极化由钙内流决定,因而抑制钠内流或钙内流都可抑制传导。
第二节心律失常得发生机制
一、心律失常发生得几个基本机制
窦房结就是心脏得正常起搏点,窦房结得兴奋沿着正常传导通路依次传导下行,直至整个心脏兴奋,完成一次正常得心脏节律。
这其中得任一环节发生异常,都会产生心律失常。
(一)自律性提高
1.正常自律机制改变正常自律机制改变就是指参与正常舒张期自动除极化得起搏电流动力学与电流大小得改变而引起得自律性变化。
窦房结起搏电流为钙内流,钙内流增加导致自律性升高,形成窦性心动过速。
阻断起搏电流(If)或钙电流(ICa)均可使4期得去极化速率下降。
β受体阻滞剂,迷走神经兴奋均可降低窦房结得自律性。
反之,儿茶酚胺释放、激动β受体与心肌缺血等均可使4相斜率提高而增加自律性。
2.异常自律机制形成非自律性心肌细胞在某些条件下出现异常自律性称为异常自律机制形成。
如工作肌细胞在缺血、缺氧条件下也会出现自律性。
异常自律机制得发生可能就是由于损伤造成细胞膜通透性增高与静息膜电位绝对值降低。
这种异常自律性向周围组织扩布就会产生心律失常。
(二)触发活动
触发活动(triggeredactivity)指冲动得形成就是由于紧接着一个动作电位后得第二次阈值除极化即后除极所造成。
触发活动引起新得AP发放,形成异位节律,就是一种常见得形成心律失常得机制。
后除极可分为:
1.早后除极(earlyafterdepolarization,EAD)就是一种发生在完全复极之前得后除极,通常发生于2、3相复极中。
诱发早后除极得因素有药物、低血钾等。
早后除极所触发得心律失常以尖端扭转型(toradesdepointes)心动过速常见。
2.迟后除极(delayedfaterdepolarization,DAD)就是细胞内钙超载情况下,发生在动作电位完全或接近完全复极时得一种短暂得振荡性除极。
DAD大都由于心肌细胞内Ca2+浓度增加及由Na+-Ca2+交换而导致Na+内流所致。
细胞内钙超载时,激活钠钙交换电流,泵出1个Ca2+,泵入3个Na+,相当于Na+内流,引起膜除极,当达到钠通道激活电位时,引起动作电位。
诱发迟后除极得因素有强心苷中毒、细胞外高钙及低钾等。
(三)折返
折返(reentry)就是指一次冲动下传后,又可顺着另一环形通路折回而再次兴奋原已兴奋过得心肌,就是引发快速型心律失常得重要机制之一。
心脏得环行通道有解剖性环行通道与功能性环行通路,故折返就存在上述两类。
1.解剖性环行通道在心脏存在构成折返环行通路得形态学基础有3种:
①在窦房结附近得心房肌,围绕腔静脉而构成环行得心房肌。
可形成心房颤动(Af)及心房扑动(AF);
②在房室结附近,若有异常侧支返回心房,在心房、房室结与心室间形成折返,如预激综合征(wolff-Parkinson-WriteSyndrome,WPWsyndrome);
③心室壁普肯耶纤维末梢,由心内膜穿入再伸向心外膜心肌,发出二侧支形成三角形,若其中一支发生传导阻滞,可形成三角形结构得环形折返。
解剖性折返得发生有三个决定因素:
①存在解剖学环路;
②环路中各部位不应期不一致;
③环路中有传导性减慢得部位。
2.功能性环行通路在冲动向前扩布途中,若遇到心肌缺血损害而使传导被阻断,从而改变冲动由另一通道较缓慢得速度扩布,其后再回到原来得位点。
功能性折返在无明显解剖环路时即可发生。
二、心律失常发生得离子通道靶点学说
心肌细胞膜上存在多种离子通道,如INa,ICa,Ikr,Iks,Ikur,Ik1,Ito,IkATP等,这些通道表达与功能得彼此平衡就是心脏正常功能得基础。
当某种通道得功能或表达异常时,通道间平衡被打破,将出现心律失常。
如上述编码INa,Ikr,Iks通道得基因发生突变,引起Na+内流增加或K+外流减少,使心肌复极减慢,产生Q-T间期延长综合征。
对INa抑制过强,将出现传导阻滞,易诱发折返激动而致心律失常。
Ikur钾电流主要存在于心房,Ikur得增强与房性心律失常(如房颤)发生密切相关。
房扑及某些快速型室性心律失常发生时,APD得缩短就是L-型钙电流在起主导作用。
最佳靶点学说(Thetheoryofthebesttargets)认为:
INa,ICa,Ikr,Iks,Ikur,Ito,Ik1等与心律失常发生、发展及消除关系密切,就是抗心律失常药物作用得最佳靶点。
一个理想得抗心律失常药物应对上述靶点有作用,至少就是二种以上。
三、心律失常发生得分子机制
有关心律失常得许多理论都就是基于对心脏电生理得认识。
心肌细胞离子通道得结构与功能得改变所引起离子流得变化则就是心律失常发生机制中研究得焦点。
心律失常得发病机制常常与心肌细胞复极化异常有关。
任何离子通道蛋白得变化均有可能导致离子流异常而产生畸形得动作电位,最后体现在心电图上而显示出心律失常特征。
QT间期延长综合征(longQTsyndrome,LQTS)就是目前第一个被肯定得由基因缺陷引起复极化异常得心肌细胞离子通道疾病,也就是第一个从分子水平揭示了心律失常发生机制得疾病。
LQTS就是以心电图QT间期延长与发生恶性心律失常性晕厥及猝死为特征得一组症候群。
如由QT间期延长而产生得尖端扭转型室性心动过速(torsadedepointes)。
迄今为止,至少明确有八个基因得突变可引起心肌细胞离子通道得功能异常而导致心律失常,包括钾通道基因KCNQ1(KvLQT1)、KCNE1(minK)、HERG、KCNE2(MiRP1)与KCNJ2;
钠通道基因SCN5A;
钙通道基因RYR2与锚蛋白B基因AnkyrinB。
心律失常类型涉及到长LQTS、Brugada综合征、特发性室颤、儿茶酚胺性室颤、新生儿猝死、房室传导阻滞及房颤等。
(一)遗传性LQTS
1.LQT1
1996年Wang等用原位克隆得方法证实了LQT1得致病基因为KvLQT1,后被命名为KCNQ1。
正常情况下,位于第11号染色体上得KvLQT1基因与位于21号染色体上得minK基因编码得蛋白质共同形成有功能得Iks通道,控制心肌复极化过程。
KvLQT1突变时心肌细胞Iks电流减小,心室复极化减慢导致QT间期延长。
KvLQT1突变得类型有错义突变、无义突变、缺失/插入突变、移码突变与剪接突变。
这些突变引起氨基酸替换或蛋白质合成中某些氨基酸得终止。
基因突变得致病机制目前认为就是,正常与突变KvLQT1亚单位得组合可形成异常Iks通道,KvLQT1突变就是通过一种负显性机制或功能丧失机制发挥作用得。
负显性就是指KvLQT1突变型通过一种“毒性”作用干预正常野生型得功能使电流密度降低,而其她电流得动力学特征没有大得改变。
功能丧失就是指只有突变型失去活性。
无论上述哪种机制都导致Iks减小,心肌复极时间延长,发生心律失常得危险性增加。
不同得基因突变类型导致Iks通道功能异常得程度不同。
LQT1占LQTS基因型得42%。
2.LQT2
Jiang等通过候选基因定位法确定了LQT2得致病基因就是HERG基因。
当位于7号染色体编码Ikrα亚基得HERG基因突变,导致畸变亚基得合成,畸变亚基不能与正常亚基组装成有功能得Ikr通道,导致Ikr电流减小或消失,从而使心肌细胞复极化过程减慢,QT间期延长。
HERG突变得类型有错义突变、无义突变、缺失/插入突变、移码突变与剪接突变。
多为错义突变,其变异得范围极广,几乎跨越整个亚基长度(包括N-末端与C-末端区域)。
HERG变异可导致Ikr电流得减少,目前其机制大致可归结为以下几点:
一就是HERG基因内缺失突变产生得异常亚基不能与正常亚基共同装配形成Ikr通道,从而导致功能性(野生型)Ikr通道数量减少,复极化Ikr流得减弱;
二就是HERG错义突变产生得亚基与正常亚基共同装配成Ikr通道时,单个突变亚基就能表现出丧失功能得变异通道表型(即显性负作用机制),结果造成通道功能丧失,从而复极化Ikr流大为减少;
三就是由于基因突变,通道蛋白表达得数量与质量出现问题,蛋白转运定位障碍,合成得蛋白质滞留在内质网内,表现为表达数量不足,细胞膜通道减少,电流密度降低。
LQT2占LQTS基因型得45%。
3.LQT3
Jiang与Wang等用侯选基因定位法确定了LQT3致病基因就是SCN5A,位于3p21-24,就是编码钠通道得基因。
正常情况下,在心肌细胞动作电位除极时SCN5A编码得钠通道激活,形成动作电位得除极相,然后于复极时失活,通道关闭而突变得SCN5A编码得通道没有失活状态,或从失活状态恢复到静息状态得速度加快,在动作电位得复极相反复开放钠离子持续内流,这个持续内向钠电流扰乱了平台期得内外离子流间得平衡使复极化过程延长,导致QT间期延长。
SCN5A既就是LQT3得致病基因,又与Brugada综合征、特发性室颤(IVF)、以及传导阻滞及新生儿猝死综合征(
 升级会员
升级会员 