第二章核磁共振氢谱Word格式文档下载.docx
《第二章核磁共振氢谱Word格式文档下载.docx》由会员分享,可在线阅读,更多相关《第二章核磁共振氢谱Word格式文档下载.docx(29页珍藏版)》请在冰豆网上搜索。
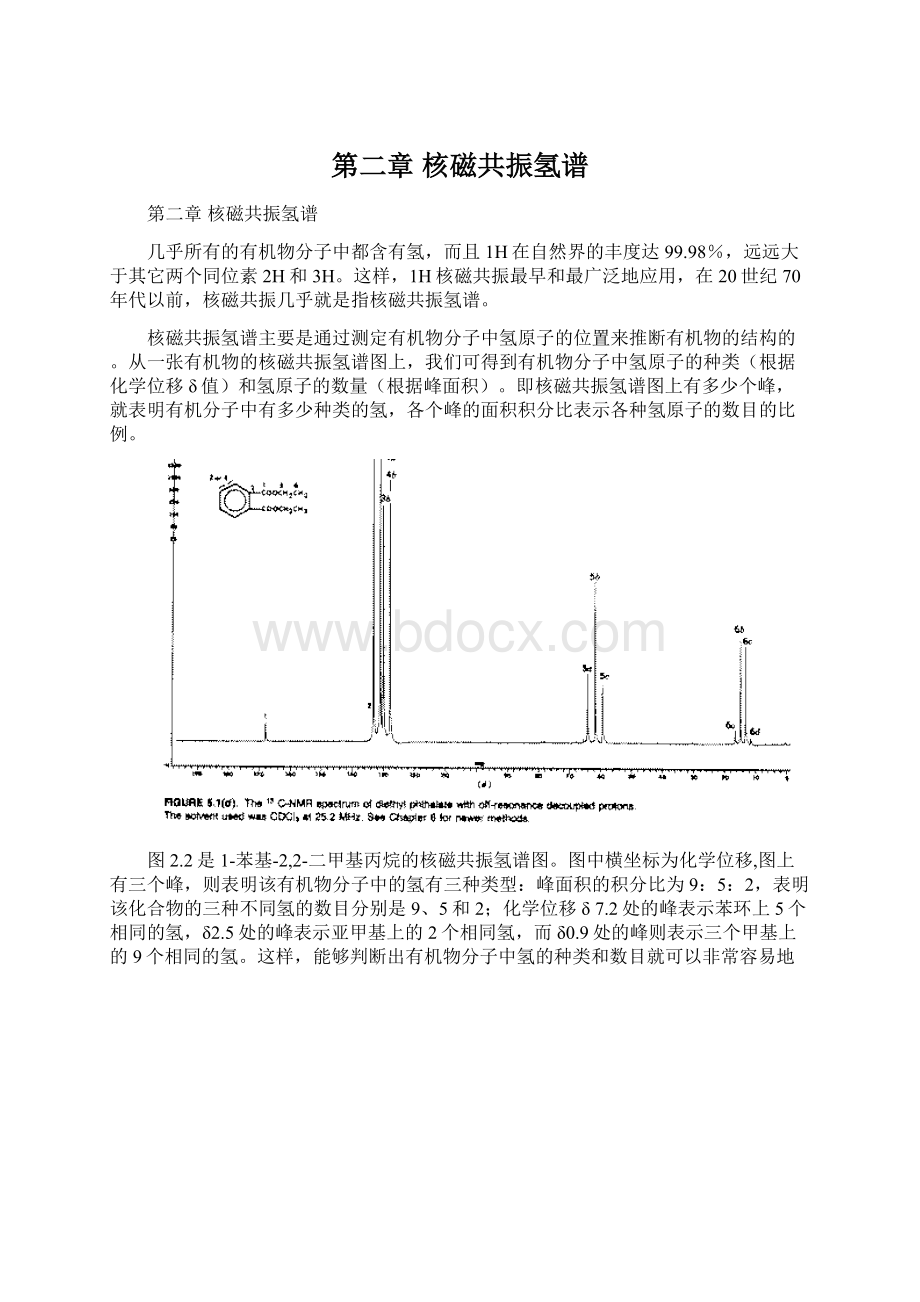
H3C-COOR
2.0-2.2
羧基取代
H3C-COOH
2.0-2.6
酰基取代
H3C-COR
2.0-2.7
炔
C≡C-H
2.0-3.0
苯基取代
2.2-3.0
醚基取代
R-O-CH3
3.3-4.0
溴取代
CH3Br
2.5-4.0
氯取代
CH3Cl
3.0-4.0
羟基取代
CH3OH
4.0-4.3
氟取代
CH3F
4.0-4.5
酰氧基取代
RCOO-CH3
3.7-4.1
胺
RNH2
1.0-5.0
醇
ROH
1.0-5.5
烯
C=C-H
4.6-5.9
苯
6.0-8.5
醛
RCHO
9.0-10.0
羧酸
RCOOH
10.5-12.0
酚
4.0-12.0
烯醇
C=C-OH
15.0-17.0
各种含氢官能团的δ值,大家要记牢,请参阅34~36表格.
对于大部分有机化合物来说氢谱的化学位移值在0-10ppm.大致可分以下几个区
0-0.8ppm
很少见,典型化合物;
环丙烷,硅烷,以及金属有机化合物。
0.8-1.5ppm
烷烃区域.氢直接与脂肪碳相连,没有强电负性取代基。
化学位移地次序CH>
CH2>
CH3.。
如果有更多的取代基化学位移移向低场。
1.5-2.5ppm羰基区域
质子相邻羰基C=O,C=Cor苯环。
3.0-4.5ppm醚区域.(同样醇,酯有CH-Ogroup.)质子直接邻氧,如果有更多的电负性取代基化学位移移向低场。
5.0-7.0ppm双键区域
.氢直接与C=C双键相连.
7.0-8.0ppm芳环质子区域.磁各向异性作用,导致芳环质子处于去屏蔽区。
同样现象发生在醛由于羰基地磁各向异性,醛质子化学位移在9-10ppm
-OHAlcohols可以出现在任何位置,谱线的性质由多重因此影响H的交换:
pH.浓度,温度,溶剂等。
一般芳环酚羟基更趋于低场。
大多数的-NHR,-NH2和醇一样,可被交换,在2-3ppm区域显示宽峰。
-CO2H可交换,象醇(>
11ppm)
化学位移的计算
某些基团或化合物的质子化学位移可以用经验公式计算.这些经验公式是根据取代基对化学位移地影响具有加和性(additivity)的原理由大量实验数据归纳总结出来的.某些情况下估算具有较高准确度,具有实用价值,而在某些场合下,虽然误差较大,但依然有参考价值.化学位移计算主要目的是:
1).对谱线进行归属;
2).为测定分子结构提供理论依据.
1.亚甲基与次甲基的δ计算
对于亚甲基可以用Shoolery公式加以计算
δ=1.25+Σσ(2-1)
式中σ为取代基的经验屏蔽常数.表中给出其数值.
表2.2Shoolery公式中的经验屏蔽常数
取代基
σ
R
-C=C-
Ph
Cl
Br
I
OH
-OR
-OPh
-OCOR
-OCOPh
NH2
NR2
NO2
SR
-CHO
-COR
-COOH
-COOR
CN
0.0
0.8
2.0
1.9
1.4
2.3
2.7
2.9
1.0
3.0
1.2
0.7
对于次甲基的δ值依然可以用Shoolery经验公式计算,但常数项改为1.5.
δ=1.50+Σσ
烯烃的化学位移计算
δC=C-H=5.25+Z同+Z顺+Z反(2.2)
Z同,Z顺,Z反分别代表相应取代基的取代参数.参阅宁永成P40`41
δ=5.25+1.08+0.18-1.02=5.49(5.56)
δ=5.25+1.38+1.18-1.02=6.79(6.81)
苯环质子化学位移的计算
取代苯环的氢化学位移可按照下式计算:
δ=7.26+ΣS(2.3)
7.26是未取代的苯环的δ值,S是取代参数.(也有书本用Zi).
2.2.偶合常数
偶合常数反映有机化合物结构的信息,特别是反映立体化学的信息.
a)自旋偶合体系及核磁共振谱图的分类
2.3.1化学等价(chemicalequivalence)
化学等价是立体化学中的一个重要观念.如果分子中两个相同原子(或两相同基团)处于相同化学环境时,它们是化学等价.化学不等价的两个基团,在化学反应中,可以反映出不同的反应速度,在光谱,波谱的测量中,可能有不同的测量结果,因而可用谱学方法来研究化学等价性.
1.考察分子各原子核相对静止状态
可用对称操作分析两个基团能否相互交换来判断两个基团(核)是否化学等价.可分为三种情况.
两个取代基完全相同,Ha,Hb可以用二次对称轴C2和对称平面相互交换.具有相同的化学位移,它们是化学等价的.
两个取代基不同,但可以用对称平面,或者二次旋转对称轴联系起来,具有相同的化学位移,它们是化学等价的.反之则是化学不等价.
2.分子内存在着快速运动
Newman投影式
常见的分子内存在有链的旋转,环的翻转.由于分子内的快速运动,一些不能通过对称操作而交换的基团有可能为化学等价,但也不是两个相同的基团就一定成为化学等价基团.
现以Newman投影来讨论分子内旋转.
RCH2-CXYZ
123
从分子旋转的角度,分子总是处于1,2,3三种构象之一,当温度升高,链的旋转速度加大,三种构象的分子逐步接近,当无论如何,Ha与Hb也不能是化学等价的.如果把R=H,三个氢是完全等价的.所以甲基的三个氢总是在同一位置.
3.前手性(prochirality)
在有机化合物中,如果与某碳原子相连的四个基团相互不等同,则是一手性中心,如果连有一对相同基团时,该碳原子则是前手性中心.一般来说前手性中心与手性中心相连,那么这一对相同的基团肯定是化学不等价.如果不与手性中心相连则用对称面原则来判断,若存在对称面,两个基团则是对映异位的.反之则是非对映异位.
4.同一碳相同二基团
a).固定在环上CH2两个氢不是化学等价的.例如;
Δδ=0.39ppm
b).单键不能快速旋转,同碳上两个基团是不等价的.
由于C-N单键具有双键性质,不能自由旋转,氮上两个甲基是化学不等价的.
C).与手性碳相连的CH2的两个氢是不等价的.
2.3.2磁等价(magneticequivalence)
两个核磁等价必须满足下列两个条件:
1.)它们是化学等价的
2.)它们对任意另外一个核的偶合常数相同(数值与符号).例如
虽然它们的化学位移是一样,但由于与顺反氟原子的偶合常数不一样.同样下面两个苯环上Ha,Ha’质子.
2.3.3自旋体系(spinsystem)
1.定义
相互偶合的核组成一个自旋体系.体系内的核相互偶合但不与体系外如何一个核偶合.在体系内部不要求一个核和它以外所有的核都偶合.例如CH3COOC2H5分别存在A3和A3X2两个自旋体系.
1.分类的原则
1).分子中化学位移相同而且对外偶合常数也相同(磁等价),用一个大写英文字母表示,如A1,A2,A3….,下标为核的数目.
2).分子中化学位移不同的核用不同的大写英文字母表示.如果核之间的化学位移之差Δν与J数值相当,用AB,ABC,ABCD….表示,如果Δν比J大许多(Δν/J>
6),用AX,AMX,AMPX…表示.
3).化学等价但磁不等价的核用AA’,BB’表示.例如
表2.3一些分子自旋体系和波谱类型
分子
自旋体系
CH4
Cl2CHCHO
CH3CH2OH
Ph-CH=CH2
A4
AX
A3X2orA3M2X
ABXorABC
AA’XX’
ABCD
H2C=CCl2
CH2F2
CH3CH2NO2
CH3CH=CH2
ClCH=CHNO2
A2
A2X2
A3X2
ABCX3
AA’BB’
AB
2.3.4核磁共振的谱图的分类
核磁谱图可分为一级谱图,=和二级谱图.
一级谱图表现满足两个条件:
1)Δν/J>
6
2)同一核组的核必须是磁等价.
一级谱图有以下特点:
1)峰的数目可用n+1规律描述.
2)峰的强度可用二项式展开系数表示.
3)从谱图中可以直接读出δ,J.峰的中心位置为δ,相邻两峰之间的距离为J.
二级谱图则没有满足以上条件,具备以上特点.
2.4一级谱图的分析
前面我们已经讨论了一级谱图的条件和特点.
2.4.1AX体系
νAνX
AX自旋体系
(1).两条谱线的中心点为化学位移
(2).两条谱线频率之差为偶合常数.
(3).四条谱线的强度相同.
2.4.2AMX体系
AMX体系解析示意图
AMX属于一级谱,但由于体系包括三个偶合常数.解析方法有特殊之处.该体系共有12条谱线,强度大体相同,分为三组,其中心位置就是相应的化学位移,谱线间隔就是偶合常数.(如图所示)
2.5高级谱图分析
1.AB体系
AB体系示意图
AB体系有四条谱线,其中两条属于“A”跃迁,另外两条属于“B”跃迁.四条谱线左右对称,.中间两条高于两边.
Δ=δA-δB
J=A1-A2=B2-B1
δA=S+1/2Δ
δB=S-1/2Δ
S=1/4(A1+A2+B1+B2)
2.4.2ABX体系
1.ABX谱图的特征及解析方法
ABX体系谱图可分AB和X两个部分.AB部分有8条谱线,可分成两组,每组都构成AB体系.谱线位置和强度完全符合AB体系的规律.X部分有6条谱线其中两条是综合峰,6条谱线的中心为νx值.
AB部分
X部分
AB体系的理论谱图
AB部分两组四重峰的1,3两线的间隔分别为R+与R-,其中间隔大R+间隔小为为R-两组四重峰的中心分别为p,q.
p-q=1/2(JAX+JBX)(2.4)
2.两组四重峰相对位置与JAX,JBX的符号关系
(1)两组不发生交盖(eclipded),JAX,JBX符号相同.
(2)两组峰交盖JAX,JBX符号可能相同,也可能相反.
(3)两组峰完全交盖,JAX,JBX符号相反,可规定JAX>
0J,JBX<
0.
(4)两组峰一组谱线发生重叠(Overlap).JBX=0或JAX,JBX符号相反.
3.ABX体系解析
1).辨认X部分和AB部分各谱线的归属和位置.
2).由X部分的中心读出δX.
3).按照AB体系解析方法求出JAB,然后取平均值.
4)分别求出两组AB四重峰的中心p和q.
2.3波谱分析方法的简化
1.提高磁场强度
(1)提高磁场的灵敏度
根据波尔兹曼的分布,随着场强H0的增强,低能态的核子数也随着增加,从而提高仪器的灵敏度。
,实际上谱仪的灵敏度(信噪比S/N)是场强的3/2次方成正比。
S/N=H03/2
(2)提高谱仪的分辨率
随着场强的增加,特别是超导磁体,可以获得很高的磁场均匀度,从而提高谱仪的分辨率。
因此使用高频率的仪器,可简化图谱。
前面我们已经讨论了Δν/J决定了谱图的复杂程度。
J的数值反映了核磁距相互作用能量的大小,它是分子本身固有的,化学位移也是不随着仪器的频率改变而发生变化。
但是Δν的确与仪器的频率成正比。
2.重水交换
如果化合物中含有与O,N,S相连的氢,在溶液中可以进行重水交换,相应的峰消失。
其顺序是OH>
NH>
SH.这样可以简化图谱。
通常溶剂是D2O.
2.化学位移试剂
我们知道:
正常的核磁共振氢谱的化学位移值在0-10ppm范围,化学位移的范围非常窄。
假如,化学位移范围被拉大,尤其是相邻重叠峰的化学位移增加,则各峰之间就可以区别分辨。
图9.8a不加位移试剂的正己醇的核磁共振氢谱
图9.8b加位移试剂的正己醇的核磁共振氢谱
含有未配对电子的金属离子具有磁性,这样在有机样品中加入金属离子配合物往往会引起试样的核磁共振峰的化学位移变化,这类能引起有机物分子核磁共振峰化学位移变化的试剂就是位移试剂。
常用的位移试剂是镧系元素铕(Eu)和镨(Pr)的三价正离子与β-二酮及其衍生物形成的配合物。
通常Eu3+的位移试剂是使有机物分子中特定氢的化学位移增加,即向高δ值方向移动;
Pr3+位移试剂则相反,是将有机物分子中特定氢的化学位移向低δ值方向移动。
因绝大多数有机物分子中氢的化学位移δ值为正数,因而Eu3+位移试剂使用比较普遍。
最常见的商品位移试剂是Eu(DPM)3(DipivalomethanatoEuropium),其对不同类型有机物分子中的特定氢分子位移的影响有显著差异(表9.2)。
Eu(DPM)3能将胺基和羟基氢的化学位移增加到100ppm以上,而对其它有机基团氢的位移分别从3ppm增加到30ppm。
对硝基和卤化物、烯类和酚等酸性有机物,位移试剂将被分解而不可用。
我们再以正己醇为例,分析位移试剂在核磁共振氢谱中的价值。
表9.2位移试剂Eu(DPM)3对不同类型有机物分子中氢化学位移的影响
有机物分子氢类型(H)
位移增加(ppm,CCl4为溶剂)*
RCH2NH2
~150
RCH2OH
~100
30~40
20~50
RCH2CHO
11~19
RCH2COR
10~17
RCH2SOR
9~11
(RCH2)2O
10
RCH2CO2R
7
RCH2CO2CH2R
5~6
RCH2CN
3~7
硝基和卤化物、烯类
RCOOH和酚
位移试剂不可用(分解)
*每摩尔有机物样品的CCl4溶液中,1摩尔Eu(DPM)3所引起的位移变化
图9.8a是不加位移试剂的正己醇的核磁共振氢谱,四个相邻的亚甲基具有差不多的化学位移,分裂的峰重叠在一起,根本不能区分。
但当加入Eu(DPM)3位移试剂后,不仅各峰的化学位移增加,而且各峰分开,分裂的多重峰也清晰可辩(图9.8b)。
这样,就可以很容易地将正己醇分子中的各种氢都能推断出来。
必须说明:
位移试剂对有机样品化学位移的变化大小与其浓度成正比,浓度大,对有机样品化学位移的影响也就大,但浓度达到一定的值后,化学位移就不再变化。
另外,位移试剂对有机物样品化学位移的影响与有机样品测定时所用的溶剂也相关,如1molEu(DPM)3在用CCl4溶解的醚样品中能使化学位移增加10ppm,但同样浓度的Eu(DPM)3能将CDCl3溶解的醚样品的化学位移增加到17~28ppm。
4双共振(doubleresonance)
双共振又叫双照射(doubleirradiation).正常的核磁共振是用单一的电磁波对有机样品进行照射,有机物分子相邻的氢核会产生自旋偶合而发生峰分裂。
对于这些能发生自旋偶合的氢核,如果再同时利用第二个电磁波照射,而且照射频率刚好与自旋氢核的频率相同,这就消除了自旋氢核间的偶合效应,相邻氢核不再有偶合现象,峰不会发生分裂,这样就使谱图简单明辩。
这一技术对核磁共振碳谱同样适用,而且现在的核磁共振碳谱都是经过去偶的,不再有峰分裂情况发生。
自旋去偶是双共振(spindecoupling)最常用的方法。
以AX体系为例,A的谱线被X分裂,但若A被照射而共振(该照射的频率为ν1),以强的功率照射X(ν2),X的核发生共振被饱和,在A核处产生的附加磁场平均为零,这消除了X核对A核的偶合作用。
AX自旋去偶示意图
2.5NOE效应(NuclearOverhausereffect)
1DNOEexperiencedbyobservingprotoniuponsaturationofthesignalofprotonjis
NOE=s/r
whereristheselectiveR1oftheobservedprotoniandsisthecrossrelaxationrate.
The1DNOEtechniquehashadastrongrevivalwhenappliedtoparamagneticmolecules.Herethesignalstobeirradiatedcanbewellseparated(hyperfineshifted),nuclearrelaxationismoreaffectedbythecouplingwiththeunpairedelectronsthanbythecouplingwithotherprotonsand,ifthemoleculeislarge,largeeffectscanbemeasuredwithoutthedrawbackofhavingallthesignalsinanarrowspectralregion.
Toremovetheeffectsofnon-perfectselectivityandoftailexcitation,themostcommonprocedureisthatofalternatingexperimentswiththesaturationpulseappliedon-resonanceandoff-resonance,andsubtractingthelatterfromtheformer.
最后,讨论一下苯环上氢的核磁共振分裂情况,苯由于产生感应磁场而导致化学位移值达到7ppm左右。
许多有机物分子中都含有苯环,因而核磁共振氢谱图上,苯环的峰非常特征,易于判断。
正常的苯环上6个氢的化学位移δ值在7.18ppm,但有机物分子中的苯环上的氢至少被一个基团取代,最多能被六个取代基取代(此时不再有苯环氢,7ppm左右无峰)。
常见的是单取代和双取
苯环单取代苯环对位双取代苯环邻位双取代苯环间位双取代
代情况,苯环单取代,苯环上还有5个氢,这5个氢在核磁共振谱图上是否发生分裂,取决于取代基X的吸斥电子和共轭等效应的大小。
若X为饱和烷基,则Ha,Ha’,Hb,Hb’和Hc五个氢没有明显的差别,在核磁共振谱图上表现为单一的峰。
但X为O、N和S等杂原子或是C=O和C=C等不饱和共轭基团,则5个氢可以分成三组,即Ha,Ha’;
Hb,Hb’和Hc,在核磁共振谱图上表现为三重峰,化学位移δ值也依据取代基X的不同而在7ppm左右波动。
对于苯环双取代,若两个取代基相同(X=X’),则对位取代苯环上的四个氢Ha=Ha’=Hb=Hb’,在核磁共振谱图上表现为单峰。
若邻位取代,苯环上四个氢分成Ha,Ha’和Hb,Hb’两类,在核磁共振谱图上表现为双峰。
若间位取代,苯环上四个氢分成Ha;
Hb,Hb’和Hc三类,而且这三类氢还会发生偶合作用,因而在核磁共振谱图上表现位三组分裂的多重峰(参见图9.5a)。
当苯环双取代的两个取代基不同(X≠X’)时,情况更为复杂。
对于对位取代,苯环上四个氢将分成Ha=Ha’和Hb=Hb’两组,而且两组氢会发生偶合作用,在谱图上表现为两组双重峰。
对于邻位和间位取代,苯环上四个氢完全不同,依据两个取代基的情况,会分成2-4组各自分裂的峰(图9.5b)。
图9.5a双硝基不同位置取代苯的核磁共振谱图
图9.5b间和对-硝基苯乙酸的核磁共振氢谱
图9.5a是双硝基不同位置取代苯的核磁共振氢谱图。
对硝基苯谱图只有单峰,表明苯环上四个氢一致。
间硝基苯谱图有三组分裂的峰,表明苯环上四个氢在两个硝基的作用下分成三组各自偶合的峰。
而邻硝基苯谱图上有一个分裂的双重峰,表明苯环上四个氢分成能偶合的两组。
顺便一提:
由于两个硝基强大的吸电子和共轭作用,苯环上氢的化学位移δ值大大增加,已达到8.0ppm以上。
图9.5b是间和对-硝基苯乙酸的核磁共振氢谱。
由于苯环上两个取代基不同,苯环上四个氢至少被分成两组。
对于对硝基苯乙酸,苯环上四个氢分成对称的两组,因而谱图上是对称的两组双峰。
而间硝基苯甲酸,苯环上四个氢不再对称,因而谱图上峰的分裂也是不规则的。
另外,硝基苯甲酸分子中除了苯环氢外,还有羧基中羟基氢和一个亚甲基氢,3.8ppm的单峰是亚甲基氢。
第三节核磁共振氢谱解析
通过上节核磁共振氢谱的介绍,我们可以通过对核磁共振氢谱的解析,来推断有机物分子的结构。
不像红外光谱通过对有机官能团的确定只能对一些简单有机物分子的结构进行推断,核磁共振氢谱通过对有机物分子中氢原子位置的判断而能直接推断出较复杂的有机物分子结构。
事实上,现在用四谱推断有机物的分子结构,都是以核磁共振谱为基础来进行的,其它的红外、紫外和质谱方法往往成为核磁共振谱推断的有机物分子结构的辅助和修正工具。
另一方面,在解析核磁共振谱图时,谱图中的每一个峰都必须找到归宿,这与红外光谱图中只解析主要峰的情况有所不同。
正因为如此,核磁共振谱图提供的有机物分子结构信息量多,只要每个峰都能找到归宿,所推断出的结构就很准确。
核磁共振氢谱图的解析,主要依据峰的化学位移δ数值大小(参见表9.1)、峰面积和峰的分裂三种情况(参见上节后部分)。
下面我们将通过对具体的核磁共振氢谱的分析来表明其在有机物分子结构鉴定种的价值。
图9.6a分子式为C6H6O2有机物的核磁共振氢谱
图9.6a是分子式为C6H6O2有机物的核磁共振氢谱。
从图中一眼就可以看出该有机物分子中只有两种不相邻的
 升级会员
升级会员 