MaterialStudio建模学习资料Word文件下载.docx
《MaterialStudio建模学习资料Word文件下载.docx》由会员分享,可在线阅读,更多相关《MaterialStudio建模学习资料Word文件下载.docx(22页珍藏版)》请在冰豆网上搜索。
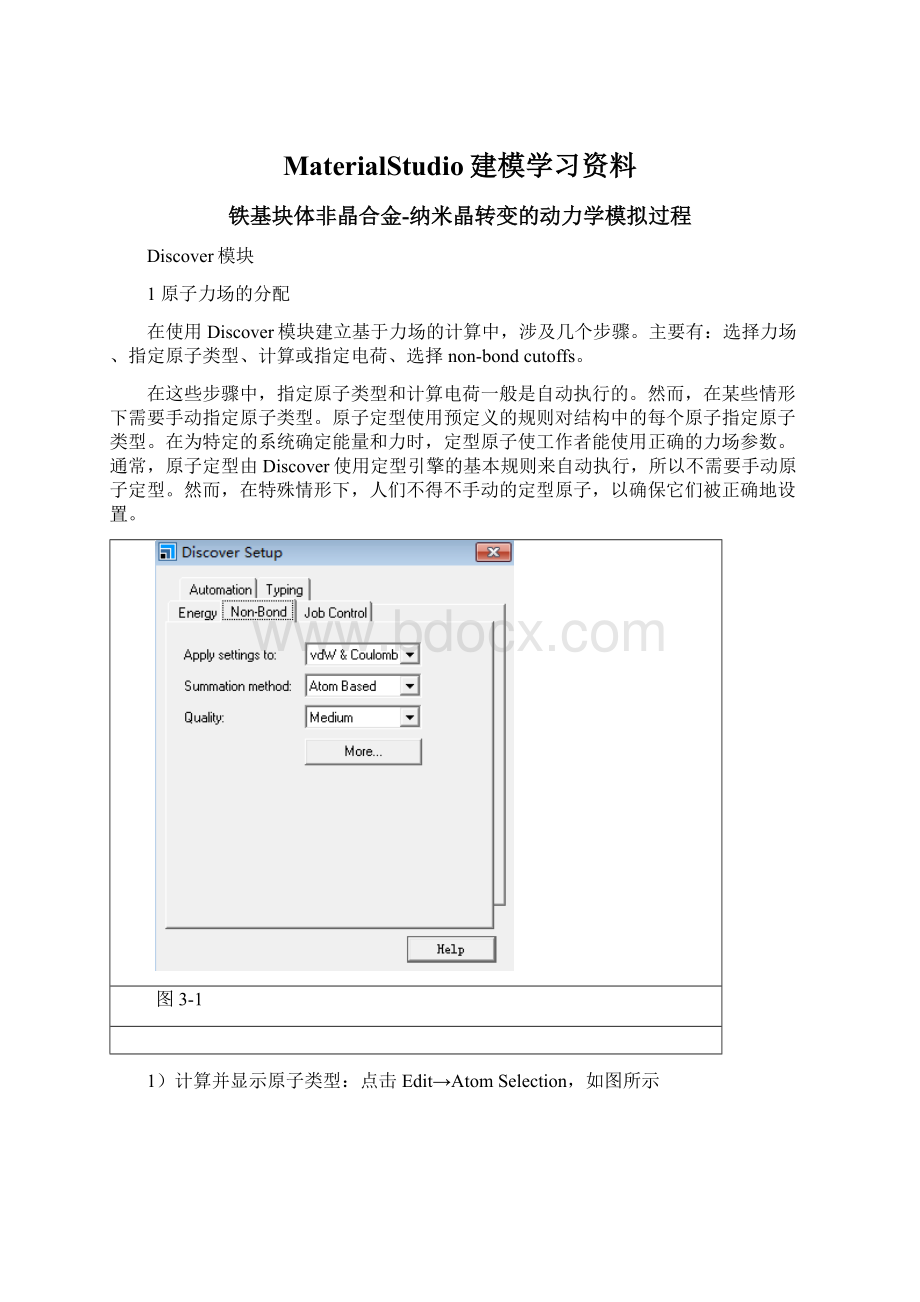
图3-1
1)计算并显示原子类型:
点击Edit→AtomSelection,如图所示
弹出对话框,如图所示
从右边的…的元素周期表中选择Fe,再点Select,此时所建晶胞中所有Fe原子都将被选中,原子被红色线圈住即表示原子被选中。
再编辑集合,点击Edit→EditSets,如图所示
弹出对话框见图,点击New...,给原子集合设定一个名字。
这里设置为Fe,则3D视图中会显示“Fe”字样,再分配力场:
在工具栏上点击Discover按钮
,从下拉列表中选择Setup,显示DiscoverSetup对话框,选择Typing选项卡。
图3-2DiscoverSetup对话框Typing选项卡
在Forcefieldtypes里选择相应原子力场,再点Assign(分配)按钮进行原子力场分配。
注意原子力场中的价态要与PropertiesProject里的原子价态(Formalcharge)一致。
2力场的选择
1)Energy
力场的选择:
力场是经典模拟计算的核心,因为它代表着结构中每种类型的原子与围绕着它的原子是如何相互作用的。
对系统中的每个原子,力场类型都被指定了,它描述了原子的局部环境。
力场包括描述属性的不同的信息,如平衡键长度和力场类型对之间的电子相互作用。
常见力场有COMPASS、CVFF和PCFF。
Select下拉菜单中有三个选项:
①COMPASS力场:
COMPASS力场是第一个把以往分别处理的有机分子体系的力场与无机分子体系的力场统一的分子力场。
COMPASS力场能够模拟小分子与高分子,一些金属离子、金属氧化物与金属。
在处理有机与无机体系时,采用分类别处理的方式,不同的体系采用不同的模型,即使对于两类体系的混合,仍然能够采用合理的模型描述。
②CVFF力场:
CVFF力场全名为一致性价力场(consistantvalenceforcefield),最初以生化分子为主,适应于计算氨基酸、水及含各种官能团的分子体系。
其后,经过不断的强化,CVFF力场可适用于计算多肽、蛋白质与大量的有机分子。
此力场以计算系统的结构与结合能最为准确,亦可提供合理的构型能与振动频率。
③PCFF力场:
PCFF为一致性力场,增加一些金属元素的力参数,可以模拟含有相应原子的分子体系,其参数的确定除大量的实验数据外,还需要大量的量子力学计算结果。
3非键的设置:
非键作用力包括范德华力和库伦力。
这里将两者都选上,为的是后期做minimizer优化原子位置时精确度更高,因为考虑了作用力因素多,即两者都考虑了。
Summationmethod(模拟方法):
1AtomBased
atombased基于原子的总量,包括一个原子的截断距离,一个原子的缓冲宽度距离;
为直接计算法,即直接计算原子对之间的非键相互作用,当原子对超出一定距离(截断半径cutoffdistance)时,即认为原子对之间相互作用为零(注:
cutoffdistance指范德瓦尔斯作用力和库仑力的范围,比如:
设定截断半径为5,则表示已分子或原子中心为圆心,以5为半径作圆,半径以外的作用力都不考虑)。
此方法计算量较小,但是可能导致能量和其导数的不连续性。
当原子对间距离在CutOff半径附近变化时,由于前一步考虑了原子对之间的相互作用,而后一步不考虑,由此会导致能量发生跳跃。
当然,对于较小的体系,则可以设置足够大的Cutoff半径来保证所有的相互作用都被考虑进来。
2GroupBased
groupbased基于电子群的,总量中包括一个原子的截断距离,一个原子的缓冲宽度距离;
大多数的分子力场都包括了每个原子之间点电荷的库仑相互作用。
甚至在电中性的物种中也存在点电荷,例如水分子。
点电荷实际上反映了分子中不同原子的电负性。
在模拟中,点电荷一般是通过电荷平衡法(chargeequilibrium)评价或者力场定义的电荷来分配的。
当评价点电荷时,一定要小心不要在使用Cutoff技术时引入错误的单极项。
要了解到这一点,可以参看如下事实:
两个单极,当只有1e.u.电荷时,在10A的位置上其相互作用大约为33Kcal;
而对于由单位单极分离1A所形成的两个偶极,相同距离其相互作用能不超过0.3Kcal/mol。
很明显,忽略单极-单极相互作用会导致错误的结果,而忽略偶极-偶极相互作用则是适度的近似。
然而,如果单极相互作用处理不清的话,仍然会出问题。
当non-bondCutoff使用基于原子-原子基组时,就可能发生,会人为将偶极劈裂为两个“假”的单极(当一个偶极原子在Cutoff内,另一个在其外)。
这就不是忽略了相对较小的偶极-偶极相互作用,而是人为引入了作用较大的单极-单极相互作用。
为了避免这种人为现象,MaterialsStudio引入了在ChargeGroups之上的Cutoff。
一个“ChargeGroup”是一个小的原子基团,其原子彼此接近,净电荷为0或者接近于0。
在实际应用中,ChargeGroup一般是常见的化学官能团,例如羰基、甲基或者羧酸基团的净电荷接近于中性ChargeGroup。
ChargeGroup之间的距离为一个官能团中心到另一个官能团中心的距离R,Cutoff设置与AtomBased相类似。
3EwaldSummation
Ewald是在周期性系统内计算Non-bond的一种技术。
Ewald是计算长程静电相互作用能的一种算法。
Ewald加和方法比较合适于结晶固体。
原因在于无限的晶格内,Cutoff方法会产生较大的误差。
然而,此方法放也可以用于无定形固体和溶液体系。
Ewald计算量较大,为o(N^3/2),体系较大时,会占用较多的内存并花费较长的时间【《分子模拟—从算法到应用》】。
4cellmultipolecellbased只能用于基于指定数量层。
一般情况下,基于Atom适合于孤立体系,对于周期性体系计算量较小,但是准确性较差;
基于Group适合于周期性和非周期性体系,计算的准确性好一些,计算量最小;
Ewald适合于周期性能体系,计算最为准确,但计算量最大。
Cutoffdistance(截断距离):
指的是范德瓦尔斯作用力和库仑力的范围。
Splinewidth:
Bufferwidth:
缓冲宽度距离。
Setup其他选项保留默认设置即可。
4结构优化
,然后选择Minimizer。
或者从菜单栏选择Modules|Discover|Minimizer。
显示DiscoverMinimizer对话框,可以进行几何结构优化计算。
注:
优化前(Min),先查看所有原子是否都已分配力场,如果没有,可以手动添加,在PropertiesExplorer中双击Forcefieldtype,然后修改力场类型即可。
其次在Min之前,需要把晶体结构所有原子重新固定。
minimizer只是对结构进行优化,以达到能量最小化。
在作动力学(dynamics)之前最好minimizer一下,因为如果不minimizer计算收敛时间会比较长,能量波动会比较大,而且计算有可能出错。
优化方法Mathod:
最陡下降法(SteepestDescent)、共轭梯度法(ConjugateGradient)、牛顿方法(Newton)和综合法(SmartMinimizer)。
Convergencelevel:
收敛精度水平。
Maximumiteration:
最大迭代数。
Optimizecell选中的话表示优化晶胞参数和原子位置。
MSDiscover结构优化原理
分子的势能一般为键合(键长、键角、二面角、扭转角等)和非键合相互作用(静电作用、范德华作用等)能量项的加和,总势能是各类势能之和,如下式:
除了一些简单的分子以外,大多数的势能是分子中一些复杂形势的势能的组合。
势能为分子中原子坐标的函数,由原子不同的坐标所得到的势能构成势能面(PotentialEnergySurface,PES)。
势能越低,构象越稳定,在系统中出现的机率越大;
反之,势能越高,构象越不稳定,在系统中出现的机率越小。
通常势能面可得到许多极小值的位置,其中对应于最低能量的点称为全局最小值(GlobalEnergyMinimum),相当于分子最稳定的构象。
由势能面求最低极小值的过程称为能量最小化(EnergyMinimum),其所对应的结构为最优化结构(OptimizedStructure),能量最小化过程,亦是结构优化的过程。
通过最小化算法进行结构优化时,应避免陷入局部最小值(localminimum),也就是避免仅得到某一构象附近的相对稳定的构象,而力求得到全局最小值,即实现全局优化。
分子力学的最小化算法能较快进行能量优化,但它的局限性在于易陷入局部势阱,求得的往往是局部最小值,而要寻求全局最小值只能采用系统搜寻法或分子动力学法。
在MaterialsStudio的Discover模块中,能量最小化算法有以下四种:
1)最陡下降法(SteepestDescent),为一经典的方法,通过迭代求导,对多变量的非线性目标函数极小化,按能量梯度相反的方向对坐标添加一位移,即能量函数的负梯度方向是目标函数最陡下降的方向,所以称为最陡下降法。
此法计算简单,速度快,但在极小值附近收敛性不够好,造成移动方向正交。
最陡下降法适用于优化的最初阶段。
2)共轭梯度法(ConjugateGradient),在求导时,目标函数下降方向不是仅选取最陡下降法所采用的能量函数的负梯度方向,而是选取两个共轭梯度方向,即前次迭代时的能量函数负梯度方向与当前迭代时的能量函数负梯度方向的线性组合。
此法收敛性较好,但对分子起始结构要求较高,因此常与最陡下降法联合使用,先用最陡下降法优化,再用共轭梯度法优化至收敛。
3)牛顿方法(Newton),以二阶导数方法求得极小值。
此法的收敛很迅速,也常与最陡下降法联合使用。
4)综合法(SmartMinimizer),该方法可以混合最陡下降法,共轭梯度法和牛顿法进行结构优化,在MS中是可选择的。
SmartMinimizer中,牛顿法可以设定最大的原子数,如果体系的原子数大于所设定的值,则计算是会自动地转为前面设定的收敛法(共轭梯度法或最陡下降法),收敛精度会改为共轭梯度法的默认收敛精度值。
点开各种方法后面的More,可设定收敛精度(Convergence),算法(Algorithm)和一维搜索(Linesearch,指每一次迭代中的精度)等。
当Job结束后,结果被返回到DiscoMin目录,最小化的结构被命名为3DAtomistic.xsd,并被保存在“3DAtomisticDiscoMin”目录。
还生成一个名为“3DAtomistic.out”的文本文档,它包含了有关计算的所有能量信息。
同时还生成“SimulationEnergies.xcd”,它显示了能量随迭代次数的变化情况。
如图所示。
本
 升级会员
升级会员 