首次注册申报资料要求及说明_精品文档Word格式.doc
《首次注册申报资料要求及说明_精品文档Word格式.doc》由会员分享,可在线阅读,更多相关《首次注册申报资料要求及说明_精品文档Word格式.doc(4页珍藏版)》请在冰豆网上搜索。
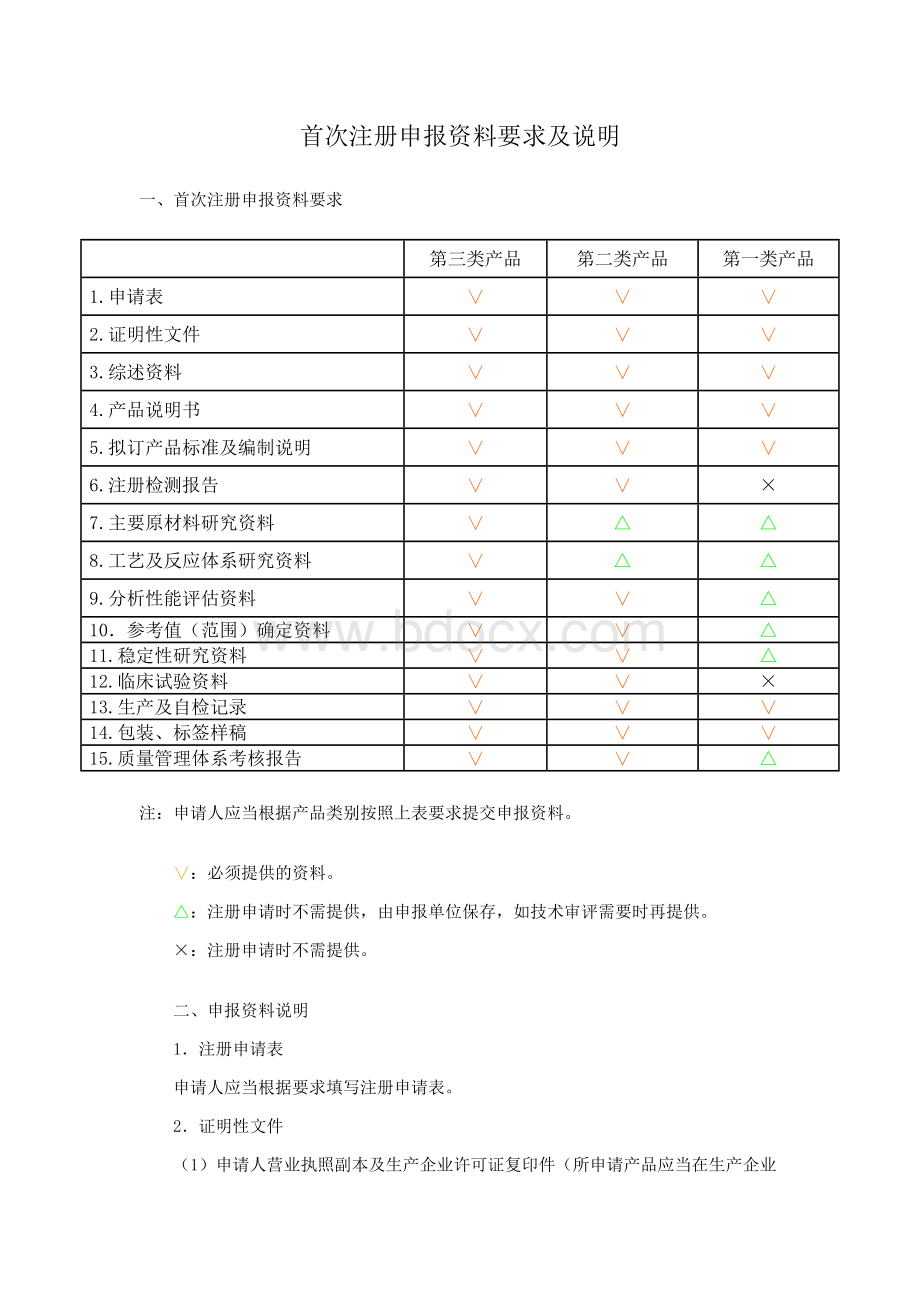
5.拟订产品标准及编制说明
6.注册检测报告
×
7.主要原材料研究资料
△
8.工艺及反应体系研究资料
9.分析性能评估资料
10.参考值(范围)确定资料
11.稳定性研究资料
12.临床试验资料
13.生产及自检记录
14.包装、标签样稿
15.质量管理体系考核报告
注:
申请人应当根据产品类别按照上表要求提交申报资料。
∨:
必须提供的资料。
△:
注册申请时不需提供,由申报单位保存,如技术审评需要时再提供。
×
:
注册申请时不需提供。
二、申报资料说明
1.注册申请表
申请人应当根据要求填写注册申请表。
2.证明性文件
(1)申请人营业执照副本及生产企业许可证复印件(所申请产品应当在生产企业许可证核定的生产范围之内)。
(2)申请人有关提交资料真实性的声明。
3.综述资料
(1)产品的预期用途:
产品的预期用途,与预期用途相关的临床适应症背景情况,如临床适应症的发生率、易感人群等,相关的临床或实验室诊断方法等。
(2)产品描述:
包括产品所采用的技术原理,主要原材料的来源及制备方法,主要生产工艺过程,质控品、标准品(校准品)的制备方法及溯源情况。
(3)有关生物安全性方面的说明:
由于体外诊断试剂中的主要原材料可能是由各种动物、病原体、人源的组织和体液等生物材料经处理或添加某些物质制备而成,为保证产品在运输、使用过程中对使用者和环境的安全,研究者对上述原材料所采用的灭活等试验方法的说明。
(4)有关产品主要研究结果的总结和评价。
(5)其他:
包括同类产品在国内外批准上市的情况。
相关产品所采用的技术方法及临床应用情况,申请注册产品与国内外同类产品的异同等。
对于新诊断试剂产品,需提供被测物与预期适用的临床适应症之间关系的文献资料。
4.产品说明书
应当符合《医疗器械说明书、标签和包装标识管理规定》的有关要求,并参考有关技术指导原则编写。
说明书中的产品名称可同时包括通用名称、商品名称和英文名称。
通用名称应当符合《体外诊断试剂注册管理办法》中有关的命名原则。
5.拟订产品标准及编制说明
拟订产品标准的文本及编制说明应当符合《医疗器械标准管理办法》的相关规定。
对于第三类产品,拟订产品标准的内容应当参照《生物制品规程》(2000版)编制。
6.注册检测报告
由国家食品药品监督管理局认可的检测机构出具的注册检测报告。
7.主要原材料的研究资料
主要原材料包括:
抗原、抗体、质控品、标准品(校准品)等的选择、制备及其质量标准的研究资料,质控品、标准品(校准品)的定值试验资料等。
8.主要生产工艺及反应体系的研究资料
主要生产工艺包括固相载体、显色系统等。
反应体系包括样本采集及处理、样本用量、试剂用量、反应条件、校准方法(如果需要)、质控方法等。
9.分析性能评估资料
(1)一般情况下,分析性能评估包括分析灵敏度、分析特异性、检测范围、测定准确性、批内不精密度、批间不精密度等项目。
分析性能评估结果是制定产品标准的重要基础。
研发者应当采用多批产品进行上述等项目的性能评估。
通过对多批产品性能评估结果进行统计分析制定拟订产品标准,以有效地控制产品生产工艺及产品质量的稳定。
如注册申请中包括不同的包装规格,或该产品适用不同机型,则需要采用每个包装规格产品,或在不同机型上进行上述项目评估的试验资料。
10.参考值(参考范围)确定:
应详细说明确定参考值(参考范围)所采用的样本来源,说明参考值(参考范围)确定的方法,并提供参考值(参考范围)确定的详细试验资料。
11.稳定性研究资料
包括至少三批样品在实际储存条件下保存至成品有效期后的稳定性研究资料,必要时应当提供加速破坏性试验资料。
12.临床试验资料
应当参考有关技术指导原则开展临床试验,并提供临床试验资料。
(1)临床试验协议及临床试验方案。
(2)各临床试验机构的临床试验报告、对所有临床试验结果的总结报告。
各临床试验机构的试验报告需加盖临床试验主管部门公章。
(3)附件:
临床试验的详细资料,包括所有临床试验结果、同时采用的其他试验方法或其他诊断试剂产品的基本信息,如试验方法、诊断试剂产品来源、产品说明书及注册批准情况等。
注:
对于校准品、质控品、参比液等不需提供临床试验资料。
13.生产及自检记录
提供连续三批产品生产及自检记录的复印件。
14.包装、标签样稿
应当符合《医疗器械说明书、标签和包装标识管理规定》的要求。
产品外包装上的标签必须包括通用名称、生产企业名称、产品批号、注意事项。
可同时标注产品的通用名称、商品名称和英文名。
对于体外诊断试剂产品中的各种组份如校准品、质控品、清洗液等,其包装、标签上必须标注该组份的中文名称和批号。
如果同批号产品、不同批号的各种组份不能替换,则既要注明产品批号,也要注明各种组份的批号。
15.质量管理体系考核报告
首次申请第三类和第二类产品注册时,应当提供相应的药品监督管理部门出具的质量管理体系考核报告。
第一类产品根据需要提供生产企业的质量管理体系自查报告。
1.上述申报资料应当由生产企业签章。
“签章”是指:
企业盖章,或者其法定代表人、负责人签名加企业盖章。
2.境内产品的注册申报资料中,如部分试验资料是由境外研究机构提供,则必须附有境外研究机构出具的有关资料项目、页码等情况的说明和证明该机构已在境外合法登记并经公证的证明文件。
但第9、11、12、13项资料必须是采用境内生产企业生产的产品所进行试验的资料。
弗锐达医疗器械技术服务有限公司
 升级会员
升级会员 