细胞生物学研究方法.docx
《细胞生物学研究方法.docx》由会员分享,可在线阅读,更多相关《细胞生物学研究方法.docx(42页珍藏版)》请在冰豆网上搜索。
细胞生物学研究方法
2.细胞生物学研究方法
生命科学是实验科学,它的很多成果都是通过实验得以发现和发展的。
方法上的突破,对于理论和应用上的发展具有巨大的推动作用。
2.1显微成像技术
最早的光学显微镜是1590年Z.Janssen和他的侄子H.Janssen共同研制的。
其后,RobertHooke和AntonievanLeeuwenhoek对光学显微镜的分辨本领进行了极大的改进,由此发现了细胞。
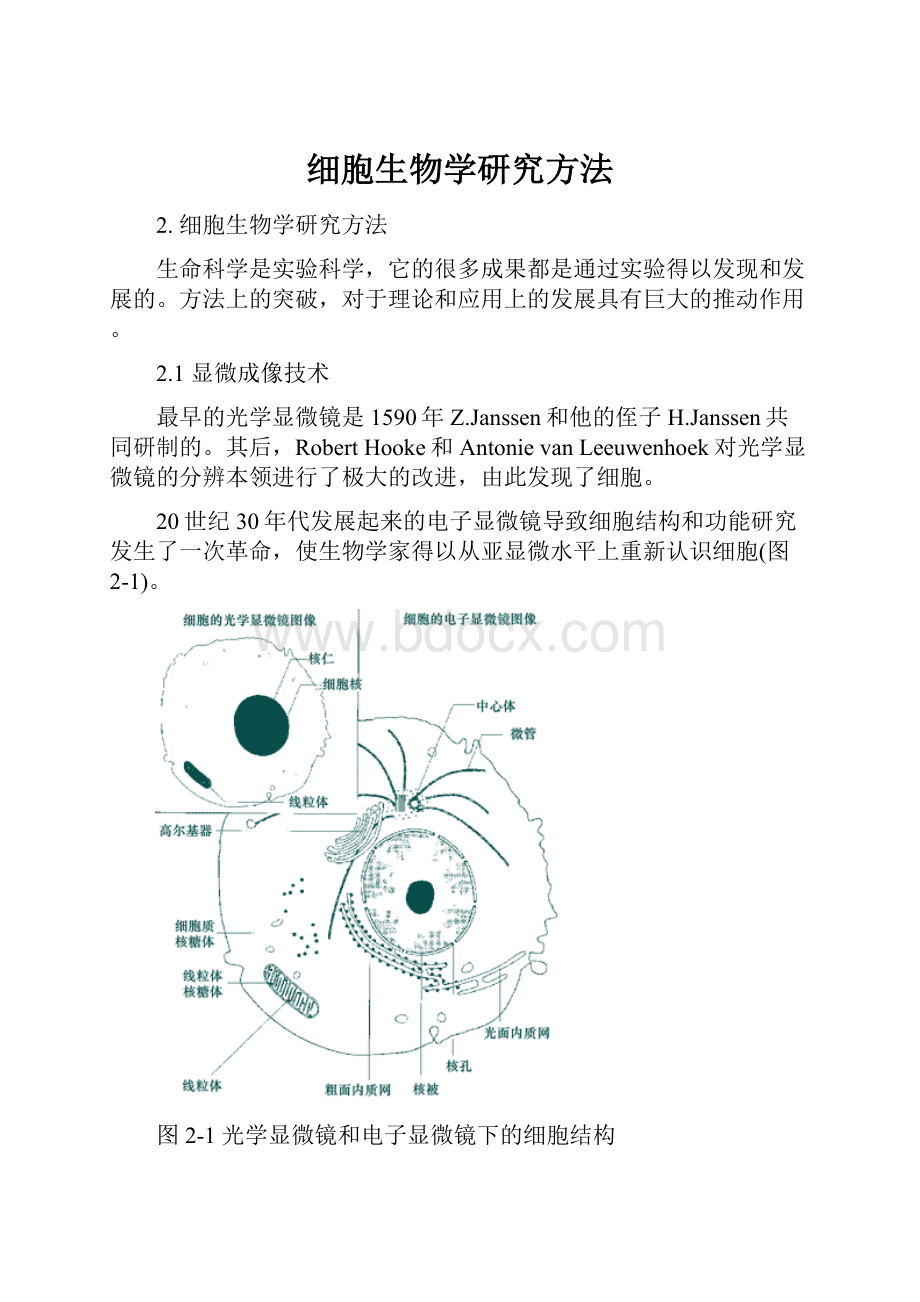
20世纪30年代发展起来的电子显微镜导致细胞结构和功能研究发生了一次革命,使生物学家得以从亚显微水平上重新认识细胞(图2-1)。
图2-1光学显微镜和电子显微镜下的细胞结构
2.1.1光学和电子显微镜成像原理
不管是何种显微镜,镜像的形需要三个基本要素:
①照明系统,②被观察的样品,③聚焦和成像的透镜系统(图2-2)。
图2-2光学和电子显微镜的基本结构
在光学显微镜中,照明系统是可见光,使用的是玻璃透镜系统,可直接通过目镜观察镜像。
在电子显微镜中,照明系统为电子束,使用电磁透镜,通过荧光屏观察样品的镜像。
照明系统的波长是显微镜成像的一个重要因素,因为波长决定能被检测样品的最小极限。
波长越长,波幅的跨度就越大,所能观察到的物体极限就越大(图2-3)。
图2-3波的移动、波长和干扰
请对图2-3作出说明
●光学和电子显微镜成像的光学原理是相同的,其中最重要的是光子和电子都具有波的行为。
当光子和电子穿过透镜到达聚焦点时,由于波的干涉(interference)性质而成像。
实际上通过透镜观察到的样品的镜像是通过透镜波的干涉累加或消除,即衍射(diffraction)的结果。
●焦距与角孔径
焦距(focallength)是透镜的中心平面到焦点的距离(图2-4),而角孔径(angularaperture)是光从样品进入显微镜的物镜半角α(图2-5),因此角孔径实际表示有多少光离开样品通过透镜,最好的光学显微镜的角孔径大约是700。
图2-4透镜的焦距
图2-5透镜的角孔径
角孔径是光从样品进入透镜的半角α。
(a)小孔径透镜;(b)大角孔径透镜。
角孔径越大,透过透镜的信息越多,最好的玻璃透镜的角孔径大约是700
■分辨率(resolution)
透镜最重要的性质就是它的分辨率,分辨率(R)可用以下公式计算:
R=0.61λ/nSinα
其中:
n=聚光镜和物镜之间介质的折射率.空气为1.油为1.5;
α=样品对物镜角孔径的半角,sinα的最大值为1;
λ=照明光源的波长。
0.61是一个恒定的参数,表示成像的点虽被重叠但仍能被区别的程度。
上式中nSinα的量称为物镜的数值孔径(numericaperture),缩写为NA,因此显微镜的分辨率的表示公式可改为:
R=0.61λ/NA
从上式可知,角孔径越大,进入物镜的光越多;介质的折射率越大,则数值孔径越大,这些都可以使分辨率提高。
由于分辨率表示的是能够区别两个点间最近距离的能力,所以R值越小,分辨率越高。
从分辨率的表达式来看,NA越大,分辨率越高,或者波长越短,分辨率越高。
■分辨极限(limitresolution)与放大率(magnification)
●一般地说,一定波长的射线不能用以探查比它本身波长短得多的结构细节,这是一切显微镜的一个基本限度。
对可见光来说,能清楚地分辨出相邻两点之间的最小间隔是0.2μm,称之为分辨极限(limitresolution)。
●最终成像的大小与原物体大小的比值称为放大率。
总放大率=物镜放大率×目镜放大率,放大率同样受分辨极限的限制。
一般来说,光学显微镜的最大放大率只能是透镜的数值孔径的1000倍。
由于透镜的数值孔径的围是1.0~1.4,所以光学显微镜在用空气作介质时最大放大倍数为1000倍,用油镜则为1400倍。
●增大角孔径或缩短波长可提高光学显微镜的分辨率。
如果用波长比普通波长短得多的电子波代替光波,分辨率可大大提高,电子显微镜就是在这种需求下被发明的。
表2-1是光学显微镜与电子显微镜某些特性的比较。
表2-2电子显微镜与光学显微镜的基本区别
分辨本领
光源
透镜
真空
光学显微镜
300nm
可见光
玻璃透镜
不需真空
200nm(油镜)
可见光
玻璃透镜
不需真空
100nm
紫外光
玻璃透镜
不需真空
电子显微镜
0.1nm
电子束
电磁透镜
真空
2.1.2常用的光学显微镜
光学显微镜(lightmicroscope)是光学显微技术的主要工具,自问世以来已有400多年历史。
光学显微镜是利用光线照明,使微小物体形成放大影像的仪器。
现今使用的光学显微镜都是由几个透镜组合而成,所以又称为复合显微镜(compoundmicroscope)(图2-6)。
图2-6普通光学显微镜的基本结构
■普通双筒显微镜(binocularmicroscope)
比较高级的显微镜上都设有倾斜式的双目镜筒(图2-7)。
在物镜转换器上方装有四个棱镜,使经过物镜的光线平分为两路到达目镜,故双筒显微镜的亮度要比单筒者为暗。
双筒显微镜的优点为同时用两眼观察,有较强的立体感。
图2-7双筒显微镜
■荧光显微镜(fluorescencemicroscope)
荧光显微镜的工作原理是利用紫外线发生装置(如弧光灯、水银灯等)发出强烈的紫外线光源,通过照明设备把显微固定的切片或活染的细胞透视出来,基本成像原理示于图2-8。
图2-8荧光显微镜的光通路
■相差显微镜(phasecontrastmicroscope)
相差显微镜在结构上进行了特别设计,尤其是光学系统有很大的不同(图2-9),可用于观察未染色的活细胞(图2-10)。
图2-9相差显微镜的光学部件及光线通路
图2-10相差显微镜观察的活细胞
■暗视野显微镜(darkfieldmicroscope)
暗视野显微镜是利用特殊的聚光器使照明光线不能进入物镜被放大,在黑暗的背景下呈现明亮的像。
这种特殊的照明方式,使反差增大,分辨率提高,用以观察未经染色的活体或胶体粒子(图2-11)。
图2-11暗视野显微镜的光学
暗视野显微镜主要观察的是物体的轮廓,分辨不清部的微细构造,适合于观察活细胞的细胞核、线粒体、液体介质中的细菌和霉菌等。
■倒置显微镜
倒置显微镜的结构组成与普通显微镜一样,所不同的只是它的物镜与照明系统的位置颠倒过来。
前者置于载物台之下,而后者在载物台的上方。
集光器与载物台之间的工作距离提高,可以放置培养皿、培养瓶等容器,直接对培养的细胞进行照明和观察(图2-12)
图2-12倒置显微镜
2.1.3光学显微镜的样品制备与观察
由于大多数细胞的成分不影响光线的穿透,无法形成反差,所以在一般光学显微镜下,几乎看不清未经处理的细胞。
为了看清细胞含物,就必须对细胞样品进行一些特殊的处理,为此建立和发展了样品的各种制备技术。
■样品的固定(fixation)
●目的:
生物组织在染色前先进行固定的目的是杀死细胞,稳定细胞的化学成份,并且使样品硬化以便在进一步的处理和切片时不会受到破坏。
●做法:
样品固定的最简单做法是将样品直接浸泡在固定液中。
固定使得大分子交联而保持在一定的位置上,不致于在以后的染色等处理过程中移位或丢失而产生人工假象。
一般用具有缓冲作用的醛类固定液,用甲醛或戊二醛作固定剂,能够与蛋白质的游离氨基形成共价键,从而将邻近的蛋白质分子牢固地交联在一起。
■包埋和切片(embeddingandsectioning)
样品制备的第二步是将固定的组织制备成切片。
为此,样品首先要被包埋在介质中,通常用液态的石蜡或树脂做包埋剂,使之渗入整块组织,然后将之硬化成固体的包埋块,随后用专门的切片机切割包埋块,制备成薄切片(图2-13)。
适用于光学显微镜观察的切片厚度为l~10μm。
图2-13用切片机进行样品切片
■染色(staining)
大多数细胞总重量的70%是水,对可见光几乎是透明的,只有很少的含物不透光。
染色的目的就是给细胞的不同组分带上可区别的颜色特征。
19世纪初,发现某些有机染料可染生物组织,并对细胞特殊部位的着色具有选择性。
如木精(hematoxylin)对负电荷分子有亲和性,能显示出细胞核酸的分布;酸性染料如伊红(eosin)可使细胞质染色;丹染料(Sudandyes)在脂肪中的溶解度比在乙醇,所以丹染料的乙醇饱和溶液能使脂肪着色。
但对许多染料的特异性染色机理尚不清楚。
■细胞化学技术(cytochemistry)
●采用比有机染料更为特异的染色剂及酶细胞化学方法,可以了解细胞和组织致的化学组成,及某些活性基团或酶的存在。
●为了测定蛋白质、核酸、多糖和脂类,常利用一些显色剂与所检测物质中特殊基团的特异性结合,通过显色剂在细胞中出现的部位和颜色显示的程度,从而判断被检物质在细胞中的分布和含量。
例如,利用Feulgen反应(图2-14)可特异性检测细胞中的DNA,PAS反应可用于检测植物中的淀粉、纤维素及动物细胞中的糖原、粘蛋白等。
●将细胞或组织切片与适宜的底物共同温育,切片中的酶会水解底物,再将所释放物质转变成不溶性有色化合物,后者所在部位即是组织细胞中酶的活性部位。
图2-14Feulgen反应
■放射自显影(autoradiography)
放射自显影技术是用感光胶片测定细胞某种被放射性标记的物质在细胞固定时所在的位置,基本过程如图2-15所示。
图2-15放射自显影术
2.1.4电子显微镜(electronmicroscope)
光源与分辨率的关系同样适于电子束,由于电子束的波长比光的波长短100,000倍,因而用电子束代替光波,可大大提高显微镜的分辨率。
1932年德国学者MaxKnolls和ErnstRuska发明了第一台电子显微镜,开拓了超微世界,发现了许多光镜下看不到的结构,如细胞膜、线粒体、细胞核、高尔基体、中心粒等细胞器的细微结构。
将在光学显微镜中观察不到而只能在电子显微镜下观察的结构称为亚显微结构(submicroscopestructure),或超微结构(ultrastructure)。
电子显微镜与光学显微镜在总体结构的设计上有很大的差别(图2-16)。
在种类上,电镜可分为两大类:
透射电子显微镜和扫描电子显微镜。
图2-16光镜与电子显微镜(透射电镜)的结构
■透射电子显微镜(transmissionelectronmicroscope,TEM)
透射电子显微镜主要是让电子束穿透样片而成像。
电子显微镜基本结构由三大部分组成∶电子光学系统(镜筒)、真空系统、电子学系统(供电系统)。
电子光学系统由照明系统、样品室、成像系统、观察窗和记录用的照相机等组成。
由于电子显微镜必需在高度真空条件下进行工作,阴极与阳极之间会放电,灯丝也会因受到氧化或被阳离子轰击而缩短寿命,所以设计了真空系统。
电子学系统即供电系统,需要高压稳压。
为什么电子显微镜需要真空系统?
■扫描电子显微镜(scanningelectronmicroscopy,SEM)
扫描电子显微镜(图2-17)使用与透射电子显微镜完全不同的方式成像,即用电视的方式成像。
扫描电子显微镜主要由电子光学系统和显示单元组成,它的光学系统结构示于图2-18。
图2-17扫描电子显微镜
图2-18扫描电子显微镜的光学系统
■扫描透射电镜与高压电镜(图2-19,2-20)
图2-19扫描透射电子显微镜
图2-20高压电镜
■电子显微镜的样品制备
如同光学显微镜,电子显微镜的样品也需固定、包埋、切片、染色等(图2-21)。
与光镜相比,用于电子显微镜的组织固定有什么特殊的要求?
图2-21电子显微镜样品的制备过程
●负染色(negativestainning)
一些生物大分子组成的结构,如病毒、线粒体基粒、核糖体和蛋白质组成的纤维等可以通过负染色电镜技术观察其精细结构,还可以从不同角度观察三维结构(图2-22)。
图2-22烟草rattle病毒的负染色
●铸型技术(shadowcasting)
铸型技术是电子显微镜中一种重要的增强背景和待观察样品反差的方法(图2-23)。
图2-23铸型技术
●冰冻断裂复型和冰冻蚀刻
是专门观察样品外表面的投影复型术(图2-24)。
图2-24冰冻断裂与冰冻蚀刻技术
2.1.5间接成像技术
光学显微镜和电子显微镜是利用质子和电子使样品直接成像的技术,还有一些显微方法是间接成像。
所谓间接成像,举一个例子,假定你拿起一个物体,非常靠近你的眼睛进行观察,你或许觉得该物体有六个平面、十二条边和八个角,然后将你感觉到的画出来,可能是一个盒子,这就是间接成像。
下面讨论的是几种间接成像的方法,这些方法都具有原子级分辨力,它们的分辨率比最好的电子显微镜还高10倍。
虽然这些方法目前都还有一定的缺陷限制着在生物学中的应用,但具有发展潜力。
■扫描隧道显微镜(scanningtunnelingmicroscope,STM)
扫描隧道显微镜由IBM公司瑞士黎世研究所的两位学者BinningG.和RohrerH.等在1981年发明的具有原子显像力的显微镜,是根据量子力学中的隧道效应原理而制成的。
这种显微镜对生物、物理、化学等学科均有推动作用,可用于研究表面的原子结构和电子结构(图2-25),故于1986年获得了诺贝尔物理学奖。
图2-25扫描隧道显微镜
DNA双螺旋结构的建立是根据X射线衍射结果推导出来的,至今还没有直接观察DNA结构的方法,所以对其中的微细结构还不够了解。
用STM观察了DNA的双螺旋结构,见到DNA分子上的大沟(majorgroove)和小沟(minorgroove),并且了解DNA的结构随染色体长度而异(图2-26)。
图2-26扫描隧道显微镜观察的DNA双螺旋结构
■X-射线衍射(X-raydiffraction)
X-衍射技术并不涉及显微镜,但是能够根据X-射线通过结晶样品形成的衍射样式成像。
这一技术可用于在原子分辨的水平上推测分子的结构。
实际上X-射线衍射技术是目前在原子水平上分析蛋白质、核酸和其他生物分子的惟一方法。
Watson和Crick提出DNA双螺旋结构模型的主要依据之一就是根据Franklin对DNA晶体衍射的结果(图2-27)。
图2-27DNA分子结构X-衍射分析
2.2细胞化学技术(cytochemistry)
细胞生物学的一个主要特点是将细胞形态观察与细胞成分分析结合起来,其中一个重要的研究手段就是细胞化学技术。
细胞化学技术不是单一的技术,而是一整套有关联的技术,包括酶细胞化学技术、免疫细胞化学技术、放射自显影技术、示踪细胞化学技术等。
2.2.1酶细胞化学技术(enzymecytochemistry)
酶细胞化学技术就是通过酶的特异细胞化学反应来显示酶在细胞的定位。
由于酶的细胞化学定位对研究细胞的生理功能和病理过程具有重要作用,而且很多酶可以作为细胞膜和各种细胞器的标志酶,这为研究细胞器的结构与功能、细胞器的相互关系以及细胞的鉴别等提供有力的手段,这一技术越来越受到重视。
2.2.2免疫细胞化学技术(immunocytochemistry)
免疫细胞化学技术是利用免疫反应定位组织或细胞中抗原成分分布的一类技术。
主要分为两大类:
免疫荧光技术和免疫电镜技术。
■免疫荧光技术(immunofluorescence)
将免疫学方法(抗原抗体特异结合)与荧光标记技术结合起来研究特异蛋白抗原在细胞分布的方法。
由于荧光素所发的荧光可在荧光显微镜下检出,从而可对抗原进行细胞定位。
■免疫电镜(immunoelectronmicroscopy)
2.2.3细胞分选技术(cellsorting)
细胞分选技术是细胞生物学研究中一个全新的技术领域,主要用流式细胞计(flowcytometer,FCM)对细胞(图2-28)或染色体(图2-29)进行分选,并进行定量分析。
■流式细胞计
流式细胞计主要由以下几部分组成∶
●激光光源:
可发出合适波长的光。
●流室(flowchamber):
生物颗粒在此与鞘液(sheathfluid)相混。
鞘液包裹着细胞的液滴经喷嘴(tip)流出,当液滴下流至适当位置,受激光照射可发射出不同的光讯号。
●讯号接受器(detector)∶由光学透镜装置和光电倍增管组成,接收放大各种光讯号,并把它们转变成电脉冲讯号。
●讯号分析部件∶为微型电子计算机装置,对讯号作出分析。
■细胞分选(cellsorting)
图2-28流式细胞计分选细胞示意图
什么是细胞分选?
原理是什么?
■染色体分选(chromosomesorting)
图2-29用于染色体分选的染色体荧光探针标记
2.2.4其他细胞化学技术
●显微分光光度术(microspectrophotometry)
●显微荧光光度术(microfluorometry)
●核磁共振技术(nuclearmagneticresonance,NMR)
.3细胞工程技术(cellengineering)
主要容包括:
细胞融合、细胞生物反应器、染色体转移、细胞器移植、基因转移、细胞及组织培养。
2.3.1细胞培养(cellculture)
在体外模拟体的生理环境,培养从机体中取出的细胞,并使之生存和生长的技术为细胞培养技术。
培养中的细胞不受体复杂环境的影响,人为改变培养条件(如物理、化学、生物等外界因素的变化)即可进一步观察细胞在单因素或多因素的影响下的生理功能变化。
体外细胞培养的条件
●物质营养
●生存环境
●废物的排除
■原代培养(primaryculture)
原代培养是指直接从机体取下细胞、组织和器官后立即进行培养。
细胞培养的一般过程如图2-30所示。
图2-30人的细胞培养
■细胞系和细胞株(celllineandcellstrain)
●细胞系:
原代培养物经首次传代成功后即为细胞系,有无限的传代能力。
●细胞株:
通过原代培养或经过细胞克隆与选择而建立的、具有特异的性质或标记的细胞系,但是它们具有有限的传代能力。
■动物细胞培养方法
●贴壁培养
分散的细胞悬浮在培养瓶中很快(几十分钟至几小时)就贴附在瓶壁上,称为细胞贴壁,贴壁后的细胞形态形成多态性,呈单层生长,所以此法又叫单层细胞培养。
单层培养的细胞保持接触抑制(contactinhibition)的特性。
●悬浮培养
悬浮培养的细胞在培养过程中不贴壁,一直悬浮在培养液中生长,如T细胞的培养就是如此。
悬浮培养的条件较为复杂,难度也大一些,但是容易同时获得大量的培养细胞。
■植物组织培养(planttissueculture)
物组织培养是根据植物细胞的全能性发展起来的利用植物植株的不同组织培养成完整植株方法。
例如叶片、茎段、根等都可以通过诱导形成愈伤组织,而后培养成植株(图2-231)。
图2-31植物组织培养
用打孔器将植物的叶片打成小圆片,然后与农杆菌进行共培养,接着放在诱导生芽的培养基上进行诱导培养,待芽长出后再转移到生根培养基上,诱导生根,最后移植到土壤中培养成完整的再生植株。
■植物原生质体培养(图2-32)
一般采用植物的体细胞(二倍体细胞),先经纤维素酶处理去掉细胞壁,这种脱去细胞壁的细胞称为原生质体。
将原生质体放在合适的培养基上,经过诱导分化可以重新长成植株。
原生质体也可用于植物细胞融合,然后诱导形成新的植株。
图2-32植物原生质体培养
2.3.2细胞融合与单克隆抗体技术
■细胞融合(cellfusion)
指自发或人工诱导下,两个不同基因型的细胞或原生质体融合形成一个杂种细胞(图2-33)。
有性繁殖时发生的精卵结合是正常的细胞融合,即由两个配子融合形成一个新的的二倍体。
图2-33细胞融合
■单克隆抗体技术(monoclonalantibodytechnique)
1975年英国科学家Milstein和Kohler发明了单克隆抗体技术,因此获得1984年诺贝尔医学奖。
图2-34所示是单克隆抗体制备流程。
一旦有了抗体就可以从事多种研究。
例如,抗体可用于蛋白质的纯化。
将抗体添加到蛋白质的粗提取液中,相应的蛋白就会同抗体结合,然后一起沉淀下来。
抗体还可用于许多免疫反应,以及应用于医学和临床。
图2-34单克隆抗体技术
■显微操作术(micromanipulation)
在显微镜下,用显微操作装置对细胞进行解剖手术和微量注射的技术属显微操作技术。
显微操作仪是在显微镜下对细胞进行显微操作的装置(图2-35),可用于细胞核移植、基因注入、染色体微切和胚胎切割等手术。
图2-35显微操作仪
2.3.3动物细胞核移植克隆技术
1997年,英国格兰罗斯林研究所I.WI.Wilmut等首次成功通过细胞融合技术利用成年动物彻底分化的体细胞克隆出子代个体,开创了动物体细胞克隆的新时代。
I.Wilmut等从成年个体母羊的乳腺组织分离单个乳腺细胞,然后与去核的羊的卵细胞融合,克隆得到一头绵羊"Dolly"(图2-36)。
体细胞克隆技术有什么意义?
图2-36细胞核移植克隆绵羊。
2.4分离技术
分离技术是一大类技术的总称,包括细胞组分的分离和生物大分子的分离。
2.4.1离心分离技术
离心分离细胞组分和生物分子是最常用的分离方法,因为不同的细胞器和分子有不同的体积和密度(图2-37),可在不同离心力的作用下沉降分离。
常用的两类离心分离方法是速度离心(velocitycentrifugation)和等密度离心(isodensitycentrifugation)。
图2-37不同的细胞器、大分子和病毒的密度及相应的沉降系数
■速度离心分离细胞器和大分子
在速度离心分离中有两种不同的方法:
●差速离心(differentialcentrifugation)(图2-38)。
图2-38差速离心的原理
●移动区带离心(moving-zonecentrifugation)(图2-39)
图2-39移动区带离心分离
将含有两种体积稍微不同的颗粒样品小心加在有轻微梯度的离心管介质的液面上(蔗糖或甘油)。
离心适当时间,样品中的颗粒向管底部移动(不能离心太久,太久了两种颗粒都会沉淀到底部),由于体积的不同,移动的区带速度不同。
然后收集不同区带的样品进行分析。
■等密度离心(图2-40)
图2-40密度梯度离心分离溶酶体、线粒体和微体
离心分离密度大于1.3g/cm3的样品,如DNA、RNA,需要使用密度比蔗糖和甘油大的介质。
重金属盐氯化铯(CsCl)是目前使用的最好的离心介质,它在离心场中可自行调节形成浓度梯度,并能保持稳定(图2-41)。
图2-41CsCl密度梯度离心分离DNA
蔗糖、甘油和氯化铯都是密度离心分离中的介质,它们在性质上、使用上和原理上有什么不同?
在速度离心时,被
 升级会员
升级会员 