休克.docx
《休克.docx》由会员分享,可在线阅读,更多相关《休克.docx(15页珍藏版)》请在冰豆网上搜索。
休克
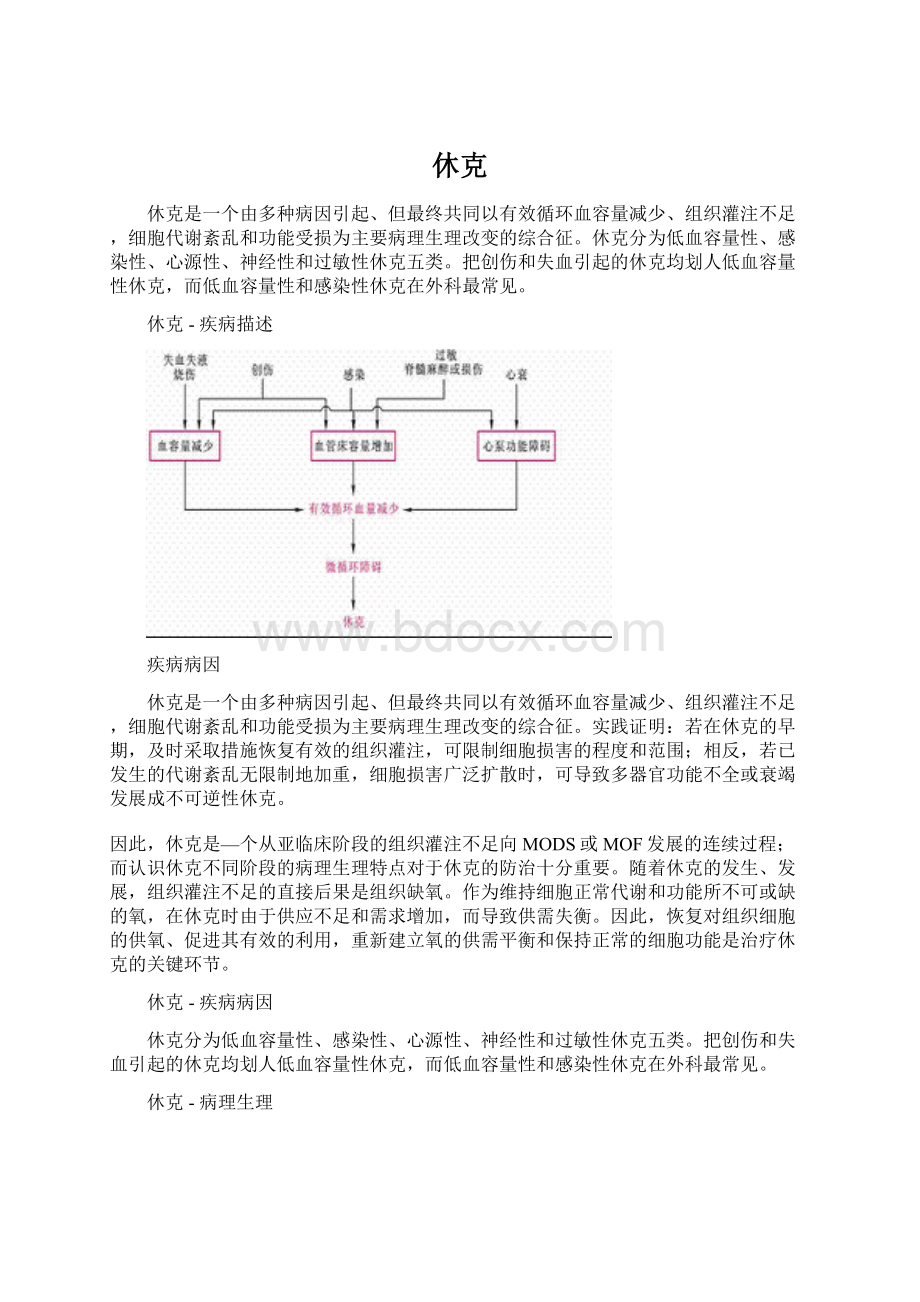
休克是一个由多种病因引起、但最终共同以有效循环血容量减少、组织灌注不足,细胞代谢紊乱和功能受损为主要病理生理改变的综合征。
休克分为低血容量性、感染性、心源性、神经性和过敏性休克五类。
把创伤和失血引起的休克均划人低血容量性休克,而低血容量性和感染性休克在外科最常见。
休克-疾病描述
疾病病因
休克是一个由多种病因引起、但最终共同以有效循环血容量减少、组织灌注不足,细胞代谢紊乱和功能受损为主要病理生理改变的综合征。
实践证明:
若在休克的早期,及时采取措施恢复有效的组织灌注,可限制细胞损害的程度和范围;相反,若已发生的代谢紊乱无限制地加重,细胞损害广泛扩散时,可导致多器官功能不全或衰竭发展成不可逆性休克。
因此,休克是—个从亚临床阶段的组织灌注不足向MODS或MOF发展的连续过程;而认识休克不同阶段的病理生理特点对于休克的防治十分重要。
随着休克的发生、发展,组织灌注不足的直接后果是组织缺氧。
作为维持细胞正常代谢和功能所不可或缺的氧,在休克时由于供应不足和需求增加,而导致供需失衡。
因此,恢复对组织细胞的供氧、促进其有效的利用,重新建立氧的供需平衡和保持正常的细胞功能是治疗休克的关键环节。
休克-疾病病因
休克分为低血容量性、感染性、心源性、神经性和过敏性休克五类。
把创伤和失血引起的休克均划人低血容量性休克,而低血容量性和感染性休克在外科最常见。
休克-病理生理
休克
有效循环血容量锐减及组织灌注不足是各类休克共同的病理生理基础。
其他与休克发生有关的病理生理过程还包括微循环改变、代谢变化和内脏器官继发性损害。
微循环的变化
在有效循环量不足引起休克的过程中,占总循环量20%的微循环也相应地发生不同阶段的变化。
休克早期,由于有效循环血容量显著减少,引起组织灌注不足和细胞缺氧;同时因循环容量降低引起动脉血压下降。
此时机体通过一系列代偿机制调节和矫正所发生的病理变化。
包括:
通过主动脉弓和颈动脉窦压力感受器引起血管舒缩中枢加压反射,交感-肾上腺轴兴奋导致大量儿茶酚胺释放以及肾素·血管紧张素分泌增加等环节,可引起心跳加快、心排出量增加以维持循环容量相对稳定;又通过选择性收缩外周(皮肤、骨骼肌)和内脏(如肝、脾、胃肠)的小直管使循环血量重新分布,保证心、脑等重要器官的有效灌注。
由于内脏小动、静脉血管平滑肌及毛细血管前括约肌受儿茶酚胺等激素的影响发生强烈收缩,动静脉间短路开放,结果外周血管阻力和回心血量均有所增加;毛细血管前括约肌收缩和后括约肌相对开放有助于组织液回吸收和血容量得到部分补偿。
但微循环内因前括约肌收蝻而致“只出不进”,血量减少,组织仍处于低灌注、缺氧状态。
若能在此时去除病因积极复苏,休克常较容易得到纠正。
若休克继续进展时,微循环将进一步因动静脉短路和直捷通道大量开放,使原有的组织灌注不足更为加重,细胞因严重缺氧处于无氧代谢状况,并出现能量不足、乳酸类产物蓄积和舒血管的介质如组胺、缓激肽等释放。
这些物质可直接引起毛细血管前括约肌舒张,而后括约肌则因对其敏感性低仍处于收缩状态。
结果微循环内“只进不出”,血液滞留、毛细血管网内静水压升高、通透性增强致血浆外渗、血液浓缩和血液粘稠度增加。
于是又进一步降低回心血量,致心排出量继续下降、心、脑器官灌注不足,休克加重而进入抑制期。
此时微循环的特点是广泛扩张。
临床上病人表现为血压进行性下降、意识模糊、发绀和酸中毒。
若病情继续发展,便进入不可逆性休克。
淤滞在微循环内的粘稠血液在酸性环境中处于高凝状态,红细胞和直小板容易发生聚集并在血管内形成傲血栓,甚至引起弥散性血管内凝血。
此时,由于组织缺少直液灌注,细胞处于严重缺氧和缺乏能量的状况,细胞内的溶酶体膜破裂,溶酶体内多种酸性水解酶溢出,引起细胞自溶并损害周围其他的细胞。
最终引起大片组织、整个器官乃至多个器官功能受损。
代谢变化
在微循环失常、灌注不足和细胞缺氧情况下,体内出现无氧代谢下的糖酵解过程以提供维持生命活动所必需的能量。
原来葡萄糖有氧代谢的开始阶段,要经糖酵解过程,1分子葡萄糖产生2分子丙酮酸;生成丙酮酸后可在脱氢酶作用下,先氧化脱羟成为乙酰辅酶A;然后进入三羧酸循环,进一步氧化成二氧化碳和水,并产生38个ATP分子,约可提供2870kJ的热量。
而无氧条件下丙酮酸只能还原成乳酸盐,产生2个ATP分子,仅提供197kJ热量,约相当于有氧代谢供能量的6.9%。
随着无氧代谢的加重,乳酸盐不断增加;丙酮酸盐则下降。
因此,在设有其他原因造成高乳酸直症的情况下,乳酸盐的含量和乳酸盐/丙酮酸盐(L/P)比值,可以反映病人细胞缺氧的情况(正常比值15~20作为缺氧的参考阈值)。
细胞微循环变化
休克加重时,除因微循环障碍不能及时清除酸性产物外,还因肝对乳酸进行代谢的能力下降,导致乳酸耸不断堆积和明显酸中毒。
当轻度酸中毒(pH>7.2)时,机体仍可受儿茶酚胺的刺激,引起心事加快、心撑出增加和血管收缩。
当发展至重度酸中毒pH<7.2时,则出现心率减慢、血管扩张和心排出量降低以及呼吸加深、加快等。
此外,酸中毒还降低心室纤颤的阈值,并使氧合直虹蛋白的解寓曲线右移,降低血红蛋白与氧的亲和力。
代谢性酸中毒和能量不足还影响细胞各种膜的屏障功能。
除了前面提到的溶酶体腆外,还影响细胞膜、核膜、线粒体膜、内质网膜、高尔基体膜等质腆的稳定及跨膜传导、运输和细胞吞饮及吞噬等功能。
细胞膜曼损后除通透性增加外,还出理细胞膜上离子系的功能障碍如Na’—K’泵、钙泵。
表现为细胞内外离子及体液分布异常,如钠、钙离于进入细胞内不能排出,钾离子则在细胞外无法进入细胞内,导致血钠降低、血钾升高,细胞外液随钠离子进人细胞内,引起细胞外液减少和细胞肿胀、死亡。
而大量钙离子进入细胞内以后除激活溶酶体外,还导致线粒体内钙离子升高,并从多方面破坏线粒体。
溶酶体腆破裂后除前面提到释放出许多引起细胞自溶和组织损伤的水解酶外,还可产生心肌抑制因子(MDF)、缓激肽等毒性因子。
线粒体膜发生损伤后,引起膜脂降解产生血栓素、白三烯等毒性产物,呈现线粒体肿胀、线粒体蜡消失,细胞氧化磷酸化障碍而影响能量生成。
能量产生不足时,影响细胞某些受体的生成。
休克-诊断检查
检查项目
一般监测
1、精神状态
是脑组织血液灌流和全身物质循环状况的反映。
例如病人神志清楚,对外界的刺激能正常反应,说明病人循环血量已基本足够;相反若病人表情淡漠,不安,谵妄或嗜睡、昏迷,反映脑因血循环不良而发生障碍。
2、皮肤温度、色泽
是体表灌流情况的标志。
如病人的四肢温暖,皮肤干燥,轻压指甲或口唇时,局部暂时缺血呈苍白,松压后色泽迅速转为正常,表明末梢循环已恢复、休克好转;反之则说明休克情况仍存在。
3、血压
维持稳定的务压在休克治疗中十分重要。
但是,血压并不是反映休克程度最敏感的指标。
例如心排出量已有明显下降时,血压的下降常滞后约40分钟;当心排出量尚未完全恢复时,血压可已趋正常,因此,在判断病情时,还应兼顾其他的参数进行综合分析。
在观察血压情况时,还要强调应定时测量、比较。
通常认为收缩压1.0-1.5有休克;>2.0为严重休克。
5.尿量
是反映肾血液灌注情况的有用指标:
尿少通常是早期休克和休克复苏不完全的表现。
对疑有休克或已确诊者,应观察每小时尿量,必要时留置导尿管。
尿量<25ml巾、比重增加者表明仍存在肾血管收缩和供血量不足;血压正常但尿量仍少且比重偏低者,提示有急性肾衰竭可能。
当尿量维持在30ml八1以上时,则休克已纠正。
此外,创伤危重病人复苏时使用高渗溶液者可能产生明显的利尿作用;涉及垂体后叶的颅脑损伤可出现尿崩现象;尿路损伤可导致少尿与无尿。
判断病情时应于注意。
特殊监测
包括以下多种血液动力学监测项目。
1.中心静脉压(CVP)
中心静脉压代表了右心房或者胸腔段腔静脉内压力的变化,在反映全身血容量及心功能状况方面一般比动脉压要早。
CVP的正常疽为0.49-0.98kPa(5-10cmH20)。
当CVP<0.49kPa时,表示血容量不足;高于1.47kPa(15cmH20)时,则提示心功能不全、静脉血管床过度收缩或肺循环阻力增高:
若CVP超过1.96kPa(20cmH20)时,则表示存在充血性心力衰竭。
临床实践中,通常进行连续测定,动态观察其变化趋势以准确反映右心前负荷的情况。
2.肺毛细血管楔压(PCWP)
应用Swan-Ganz飘浮导管可测得肺动脉压(PAP)和肺毛细血管楔压(PCWp),可反映肺静脉、左心房和左心室压。
PAP的正常值为1.3—2.9kPa(10—22nmaHg)~PCWP的正常值为0.8-2kPa(6-15mmHg)。
与左心房内压接近。
PCWP低于正常值反映血容量不足(较CVP敏感);PCWP增高常见于肺循环阻力增高例如肺水肿时。
因此,临床上当发现PCWp增高时.即使CVP尚属正常.也应限制输液量以免发生或加重n,水肿。
此外,还可在作PCWp时获得血标本进行混合静脉血气分析,了解肺内动静脉分流或肺内通气/灌流比的变化情况。
但必须指出.肺动脉导管技术是一项有创性检查,有发生严重并发症的可能(发生率约3%-5%),故应当严格掌握适应证。
3.心排出量(CO)和心脏指数(C1)
CO是心率和每搏排出量的乘积.可经Swan·Ganz导管应用热稀释法测出。
成人CO的正常值为4—6L/min;单位体表面积上的心排出量便称作心脏指数(C1),正常值为2.5—3.5L/(min‘m2)。
此外,还可按下列公式计算出总外周血管阻力(SVR):
SVR=(平均动脉压-中心静脉压)/心排量×80心搏出量正常值为100—130kPa·s/L了解和检测上述各参数对于抢救休克时及时发现和调整异常的血液动力学有重要意义。
通常在休克时,CO值均较正常值有所降低;有的感染性休克时却可能高于正常值。
因此在临床实践中,测定病人的CO值并结合正常值进行调整固然必要,但更重要的是结合具体病情确定一个在病理情况下既满足代谢需要,又不增加心血管负荷、对每个具体病人最适宜的CO值。
这对治疗心源性休克尤其重要。
适宜心排出量的确定,可用带有分光光度血氧计的改良式肺动脉导管,连续测定混合静脉血氧饱和度(SvO2),来判断体内氧供应与氧消耗的比例。
反映正常人体内氧供应与消耗之间达到平衡的SvO2值是0.75。
SvO2值降低则反映氧供应不足,可因心排出量本身降低、血红蛋白浓度或动脉氧饱和度降低所致。
此外,确定适宜的CO还可经动态地观察氧供应和氧消耗(V02)的关系来判断。
先在原来的CO情况下通过强心、扩容措施,逐渐地提高D02,观察V02的反应。
当V02随D02而相应提高时,称作“氧供依赖性氧耗”,反映DO2不能满足机体代谢需要,提示应继续努力提高CO以免发生机体缺氧,直至V02不再随D02升高而增加为止。
即使此时CO值仍低于正常值,也表明D02已满足机体代谢需要。
休克
4.动脉血气分析
动脉血氧分压(PaO2)正常值为10.7—13kPa(80—100illmHg);当降至4kPa时,组织便已处于无氧状态。
动脉血二氧化碳分压(PaC02)正常值为4.8—5.8kPa(36--44mmHg)。
休克时可因肺换气不足,出现体内二氧化碳聚积致PaC02明显升高;相反,如病人原来并无肺部疾病,因过度换气可致P~032较低;若病人通气良好,但PaC02仍超过5.9-6.6kPa(45—50mmHg)时,常提示严重的肺泡功能不全;PaC02高于8.0kPa(60mmHg),吸人纯氧仍无改善者则可能是ARDS的先兆。
动脉血pH正常为7.35~7.45。
通过监测pH、碱剩余(BE)、缓冲碱(BB)和标准重碳酸盐(SB)的动态变化有助于了解休克时酸碱平衡的情况。
5.动脉直乳酸盐测定
休克病人组织灌注不足可引起无氧代谢和高乳酸血症,监测有助于估计休克及复苏的变化趋势。
正常值为1-1.5mmol几,危重病人允许到2hanoi/L。
此外,还可结合其他参数判断病情,例如乳酸盐/丙酮酸盐(L/P)比值在无氧代谢时明显升高;正常比值约10:
1,高乳酸血症时L/P比值升高。
6.DIC的检测
对疑有D1C的病人,应测定其血小板的数量和质量、凝血因子的消耗程度及反映纤容溶活性的多项指标。
当下列五项检查中出现三项以上异常,结合临床上有休克及微血管栓塞症状和出血倾向时,便可诊断DIC包括:
①血小板计数低于80xIO~/L;②凝血酶原时间比对照组延长3秒以上;①血浆纤维蛋白原低于1.5g/L或呈进行性降低;④3P(血浆鱼精蛋白副凝)试验阳性和⑤血涂片中破碎红细胞超过2%等。
7.胃肠粘膜内pH(phi)值监测
根据休克时胃肠道较早便处于缺血、缺氧状态,因而易于引起细菌移位、诱发脓毒症和MODS;而全身血液动力学检测常不能反映缺血严重器官组织的实际情况。
测量胃粘膜pHi,不但能反映该组织局部灌注和供氧的情况,也可能发现隐匿性休克。
phi测定是用间接方法:
首先经鼻向胃内插入带半透膜囊腔的胃管,向囊腔注入4ml盐水,30一90分钟后测定该盐水中的PCO2;同时取动脉血,用血气机测出HCO3和PCO2;然后将胃管内的盐水PCO2与动脉血HCO3值代入下列公式算出ph值:
phi=6.1十log(动脉HC03/33x胃管生理盐水PC02)pHi的正常范围为7.35—7.45。
休克-休克并发症
可发生心力衰竭、急性呼吸衰竭、急性肾功能衰竭、脑功能障碍和急性肝功能衰竭等并发症。
1、按病因分类
(7种):
失血性、烧伤性、创伤性、感染性、过敏性、心源性、神经源性。
2、按休克的起始环节分类
(正常时保证微循环有效灌注的基础因素包括三方面:
足够的循环血量、正常的血管容量、正常的心泵功能):
(1)低血容量性休克见于失血、失液或烧伤等,血容量减少导致静脉回流不足,心输出量下降,血压下降,由于减压反射受抑制,交感神经兴奋,外周血管收缩,组织灌流量进一步减少。
(2)血管源性休克休克时,由于组织长期缺血、缺氧、酸中毒和组胺及一氧化碳等活性物质的释放,造成血管张力低下,加上白细胞、血小板在微静脉端黏附,造成微循环血液瘀滞,毛细血管开放数增加,导致有效循环血量锐减。
a、过敏性休克:
属于1型变态反应。
发病机制与IgE及抗原在肥大细胞表面结合,会引起组胺和缓激肽大量入血,造成微动脉扩张,微静脉收缩,微循环瘀血,毛细血管通透性增加。
b、感染性休克:
(常伴败血症) ①高动力型休克:
由于扩血管因子作用大于缩血管因子,引起高拍低阻的血流动力学特点。
②低动力型休克。
c、神经源性休克:
由于麻醉或损伤和强烈的疼痛抑制交感神经缩血管功能,引起一过性的血管扩张和血压下降, 此时微循环不一定明显减少,有人认为不属于真正的休克。
(这句话不很明白,抄来的,有人可以请教吗?
真正的休克本质上是微循环障碍,这种神经源性的休克只是引起一过性的血管扩张而血压降低,尚未导致微循环出问题-by yukimiao)
(3)心源性休克 发病中心环节时心输出量迅速降低,血压可显著下降。
(低动力型休克) ①低排高阻型:
是因为血压降低,主动脉弓和颈动脉窦的压力感受器的冲动减少,反射性引起交感神经传出冲动增多,引起外周小动脉收缩,使血压能有一定程度的代偿。
②低排低阻型:
这类病例是由于心肌梗死面积大,心输出量显著降低,血液瘀滞在心室,使心室壁牵张感受器受牵拉,反射性地抑制交感中枢,使交感神经传出冲动减少,外周阻力降低,引起血压进一步减少。
休克-危害
1、肺:
休克时的缺血、缺氧,可使得肺部的毛细血管、肺泡细胞受到损伤。
而且在治疗休克时还可引起肺部小血管栓塞,使部分肺泡萎陷、不张、水肿,部分血管闭塞、不通畅,结果就是流入肺部的血液不能很好的从肺得到氧气,各器官也就供氧不足。
严重时可导致急性呼吸窘迫综合征。
2、肾:
病人处于休克状态时,肾脏就不能得到足够的血液供应,严重时可导致肾脏缺血坏死,甚至出现急性肾功能衰竭。
3、脑:
脑部血流量减少,可导致脑组织缺血、缺氧,而缺血、缺氧又可引起脑细胞肿胀、血管通透性升高,从出现脑水肿和颅内压增高。
这时的病人可出现意识障碍,严重者可发生脑疝、昏迷。
4、心:
休克时冠状动脉血流减少,从而心肌缺血,最后引起心脏肌细胞损伤。
5、胃肠道:
休克可引起胃肠道溃疡、肠源性感染。
6、肝:
休克可引起肝缺血、缺氧性损伤,肝脏功能障碍。
重要器官功能衰竭
(1)急性肾功能衰竭——休克肾
①功能性肾功能衰竭:
见于休克早期,主要与各种缩血管物质增多使肾血管收缩有关。
因未发生肾小管坏死,肾血流一旦恢复,肾功能也容易逆转。
②器质性肾功能衰竭:
见于休克期,尤其是休克晚期,由于长时间缺血和毒素的作用可造成肾小管坏死,即使肾血流恢复,也较难在较短的时间内恢复肾功能。
(2)急性呼吸功能衰竭——休克肺(ARDS之一)
发生机制:
①肺泡-毛细血管上皮通透性增高:
由于休克致病因子的直接作用和多种细胞因子的间接作用,可使肺泡-毛细血管膜损伤、通透性增高,引起渗出性肺水肿。
②肺泡表面活性物质减少:
缺血缺氧使肺泡Ⅱ型上皮细胞受损,以致表面活性物质合成减少;同时肺泡腔的水肿液可加速表面活性物质的分解,结果是肺泡表面张力增高,肺顺应性降低引起肺不张。
③肺内DIC:
DIC造成肺微血管的机械阻塞 以及来自微血栓的炎症介质对肺血管的收缩可导致死腔样通气。
(3)脑功能障碍
休克早期脑供血未明显改变,患者表现为烦躁不安;休克期因脑供血减少,患者出现神志淡漠;休克晚期可因DIC而导致昏迷或意识丧失。
(4)胃肠道和肝功能障碍
①胃肠功能障碍:
休克时胃肠因缺血、瘀血及DIC形成,使消化液分泌减少及胃肠蠕动减弱,消化功能明显障碍;持续的缺血,不仅可致胃粘膜糜烂而发生应激性溃疡,还可因肠道屏障功能受损和细菌的大量繁殖导致全身炎症反应综合征。
②肝功能障碍:
休克时肝脏缺血、瘀血可发生肝功能障碍,由于不能将乳酸转化为葡萄糖,可加重酸中毒;尤其来自肠道的内毒素可直接损伤肝细胞,从而促进休克的发展。
(5)心脏
发生机制:
①冠脉供血减少:
休克时血压下降以及心率过快引起的心室舒张时限缩短,可使冠脉灌注减少。
②酸中毒和高钾血症使心肌收缩性减弱。
③心肌抑制因子抑制心肌收缩性。
④心肌内DIC使心肌受损。
⑤细菌毒素,尤其内毒素可直接损坏心肌。
休克-治疗方案
药物治疗
对于休克这个由不同原因引起、但有共同临床表现的综合征,应当针对引起休克的原因和休克不同发展阶段的重要生理紊乱采取下列相应的治疗。
一般紧急治疗
包括积极处理引起休克的原发伤病。
如创伤制动、大出血止血、保证呼吸道通畅等。
采取头和躯干抬高20·—30·;下肢抬高15·一20·体位,以增加回心血量。
及早建立静脉通路。
并用药维持血压。
早期予以鼻管或面罩吸氧,注意保温。
补充血容量
是纠正休克引起的组织低灌注和缺氧的关键。
应在连续监测动脉血压、尿量和CVP的基础上,结合病人皮肤温度、末梢循环、脉搏幅度及毛细血管充盈时间等微循环情况,判断补充血容量的效果。
通常首先采用晶体液,但由于其维持扩容作用的时间仅1时左右.故还应准备全血、血浆、压缩红细胞、清蛋白或血浆增量剂等胶体液输注。
也有用3%一7.5%高渗盐溶液行休克复苏治疗。
通过高溶液的渗透压作用,能吸出组织间隙和肿胀细胞内的水分起到扩容的效果;高钠还有增加碱储备和纠正酸中毒的作用。
成人烧伤面积在15%,小儿10%以上时要进行抗休克补液。
补液公式:
成人:
烧伤每百分之一体表面积补充晶胶体1.5ml/kg,水分3000ml,尿量维持在50~70ml/h,小儿每百分之一补充晶胶体1.0ml/kg,水分约100ml/kg,尿量维持在0.5~1.0ml/kg/h。
积极处理原发病
外科疾病引起的休克,多存在需手术处理的原发病变,如内脏大出血的控制、坏死肠袢切除、消化道穿孔修补和脓液引流等。
应在尽快恢复有效循环血量后,及时施行手术处理原发病变,才能有效地治疗休克。
有的情况F.应在积极抗休克的同时进行手术,以免延误抢救时机。
纠正酸碱平衡失调
休克病人由于组织灌注不足和细胞缺氧常有不同程度的酸中毒,而酸性内环境对心肌、血管平滑肌和肾功能均有抑制作用。
在休克早期,又可能因过度换气,引起低碳酸血症、呼吸性碱中毒。
按照血红蛋白氧合解离曲线的规律,碱中毒使血虹蛋白氧离曲线左移,氧不易从血红蛋白释出.可使组织缺氧加重。
故不主张早期使用碱性药物。
而酸性环境有利于氧与血红蛋白解离,从而增加组织供氧,机体在获得充足血容量和微循环改善后,轻度酸中毒常可-缓解而不需再用碱性药。
但重度休克合并酸中毒经扩容治疗不满意时,仍需使用碱性药物。
用药前需保证呼吸功能正常,以免引起Cq潴留和继发呼吸性酸中毒。
常用的碱性药物及用量见第三章,给药后应按血气分析的结果调整剂量。
血管活性药物的应用
严重休克时,单用扩容治疗不易迅速改善循环和升高血压。
若血容量已基本补足但循环状态仍未好转表现发绀、皮肤湿冷时,则应选用下列血管活性药物:
1.血管收缩剂
有去甲肾上腺素、间羟胺和多巴胺等。
去甲肾上腺素是以兴奋。
—受体为主、轻度兴奋β-受体的血管收缩剂,能兴奋心肌.收缩血管,升高血压及增加冠状动脉直流量,作用时间短。
常用量为0.5-2mg,加入5%葡萄糖溶液100nd内静脉滴注。
间羟胺(阿拉明)间接兴奋α和β受体,对心脏和血管的作用同去甲肾上腺素,但作用弱,维持时间约30分钟。
常用量2-10mg肌注或2-5mg静脉注射;也可10~20mg加入5%葡萄糖溶液100mi静脉滴注。
多巴胺是最常用的血管收缩剂,具有兴奋。
α、β和多巴胺受体作用,其药理作用与剂量有关。
小剂量[15ug/(min))时则为α受体作用,增加外周血管阻力。
抗休克时主要取其强心和扩张内脏血管的作用,宜采取小剂量。
为提升血压,可将小剂量多巴胺与其他缩血管药物合用,而不增加多巴胺的剂量。
多巴酚丁胺对心肌的正性肌力作用较多巴胺强,能增加120,降低CWP,改善心泵功能。
常用量为2.5~10~g/(kg·mm)。
小剂量有轻度缩血管作用。
异丙基肾上腺素是能增强心肌收缩和提高心率的β受体兴奋剂,剂量0.1—0.2mg溶于100ml输液中。
因对心肌有强大收缩作用和容易发生心律紊乱,不能用于心源性休克。
2.血管扩张剂
分受体阻滞剂和抗胆碱能药两类。
前者包括酚妥拉明、酚苄明等,能解除去甲肾上脓素所引起的小血管收缩和微循环淤滞并增强左室收缩力。
其中酚妥拉明作用快,持续时间短,剂量为0.1-0.5rng/kg加于100ml静脉输液中。
酚苄明是一种α-受体阻滞剂,兼有间接反射性兴奋β-受体的作用。
能轻度增加心脏收缩力、心排出量和心事,同时能增加冠状动脉血流量,降低周围循环阻力和血压。
作用可维持3—4天。
用量为0.5~1.0mg&g,加入5%葡萄糖溶液或0.9%氯化钠溶液内,l一2小时滴完。
抗胆碱能药物包括阿托品、山莨菪碱和东莨菪碱。
临床上较多用于休克治疗的是山莨菪碱(人工合成品为654-2),可对抗乙酰胆碱所致平滑肌痉挛使血管舒张,从而改善微循环。
还可通过抑制花生四烯酸代谢,降低白三烯、前列腺素的释放而保护细胞,是良好的细胞膜稳定剂。
尤其是在外周血管痉挛时,对提高血压、改善微循环、稳定病情方面,效果较明显。
用法是每次10mg,每15分钟一次,静注,或者40~80mg/h持续泵入,直到临床症状改善。
硝蕾钠也是一种血管扩张剂,作用于血管平滑肌,能同时扩张小动脉和小静脉,但对心脏无直接作用。
静脉用药后可降低前负荷。
剂量为100ml液体中加入5—10mg静脉滴注。
滴速应控制在20—100ug/min,以防其中的高铁高于转变为亚铁离子。
用药超过3天者应每日检测血硫氰酸盐浓度,超过12.8%时即应停药。
手术治疗
3.强心药
包括兴奋α和β肾上腺素能受体兼有强心功能曲的药物,如多巴胺和多巴酚丁胺等,其他还有强心甙如西地
 升级会员
升级会员 