硫酸亚铁铵的制备及组成测定Word下载.docx
《硫酸亚铁铵的制备及组成测定Word下载.docx》由会员分享,可在线阅读,更多相关《硫酸亚铁铵的制备及组成测定Word下载.docx(10页珍藏版)》请在冰豆网上搜索。
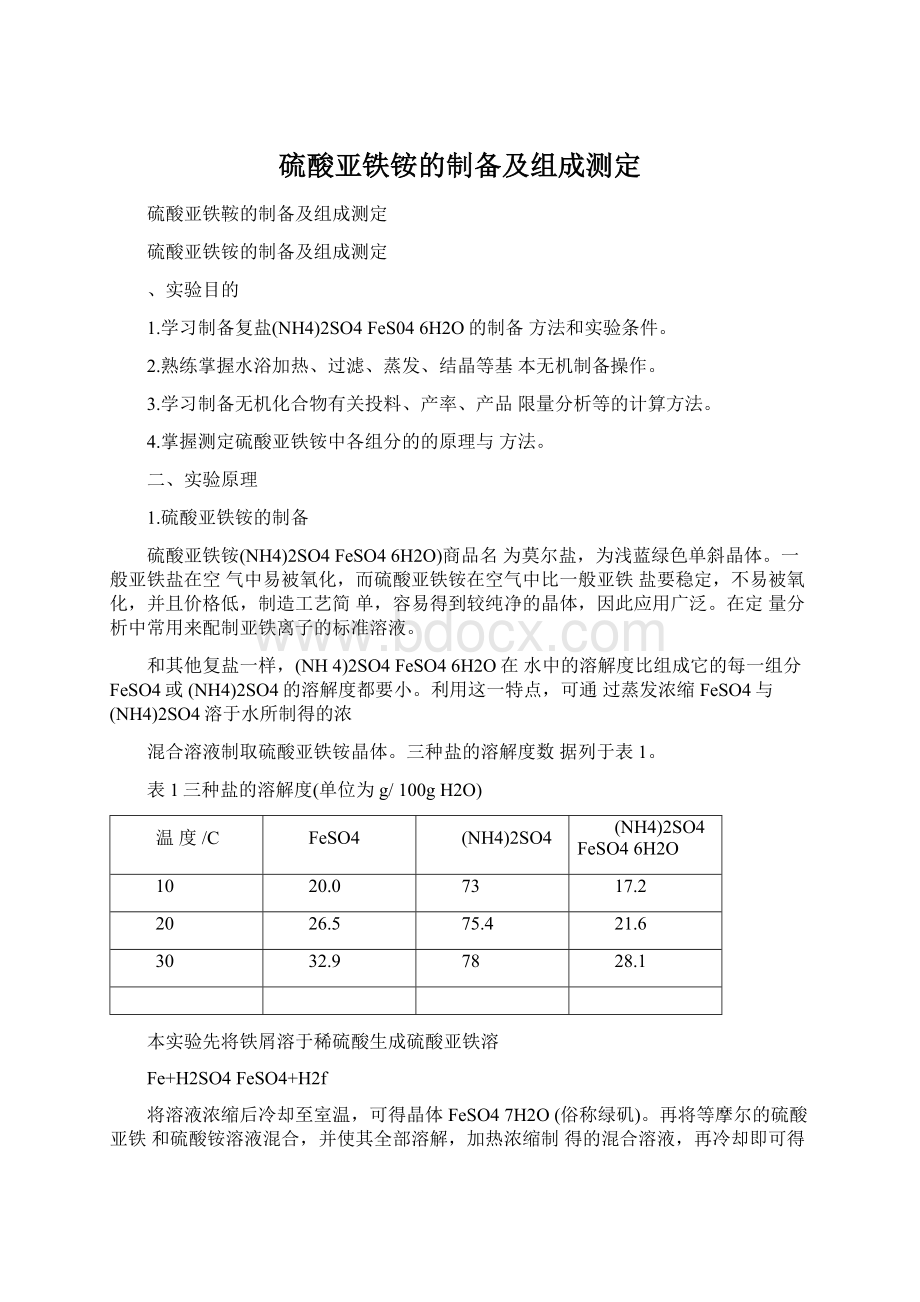
78
28.1
本实验先将铁屑溶于稀硫酸生成硫酸亚铁溶
Fe+H2SO4FeSO4+H2f
将溶液浓缩后冷却至室温,可得晶体FeSO47H2O(俗称绿矶)。
再将等摩尔的硫酸亚铁和硫酸铵溶液混合,并使其全部溶解,加热浓缩制得的混合溶液,再冷却即可得到溶解度较小的硫酸亚铁铵晶体,它的氧化还原稳定性比一般的亚铁盐高,常作为亚铁试剂使用。
FeS04+(NH4)2SO4+6H2O
用目视比色法可估计产品中所含杂质Fe3+的
量。
Fe3+与SCN—能生成红色物质[Fe(SCN)]2+,红色深浅与Fe3+相关。
将所制备的硫酸亚铁铵晶体与KSCN溶液在比色管中配制成待测溶液,将它所呈现的红色与含一定Fe3+量所配制成的标准[Fe(SCN)]2+溶液的红色进行比较,确定待测溶液中杂质Fe3+的含量范围,确定产品等级。
2.Fe2+含量测定
(1)重铬酸钾法测铁,是铁矿中全铁量测定的标准方法。
在酸性溶液中,Fe2+可以定量地被K2C“O7氧化成Fe3+,反应为:
6Fe2++Cr2O72「+14H+=6Fe3++
2Cr3++7H2O
滴定指示剂为二苯胺磺酸钠,其还原态为无
色,氧化态为紫红色。
必须加入磷酸或氟化钠等,目的有两个:
一是与生成地Fe3+形成配离子
[Fe(HPO4)]+,降低Fe3+/Fe2+电对的电极电势,扩大滴定突跃范围,使指示剂的变色范围在滴定的突跃范围之内;
二是生成的配离子为无色,消除了溶液中Fe3+黄色干扰,利于终点观察。
(2)采用邻菲啰啉比色法测定亚铁含量
在一定酸度条件下,试液中亚铁离子(Fe2+)与1,10-邻菲啰啉生成红色配合物,于波长为506nm处,测定其吸光度,即可计算出铁含量。
3.NH4+的测定
(1)甲醛法
由于铵盐中NH4+的酸性太弱,Ka=5.6X10-7(pH=6.3)。
故无法用NaOH标准溶液直接滴定。
因此,工业生产中普遍采用甲醛法测定铵盐中的含氮量。
将铵盐与甲醛作用,定量生成六亚甲基四铵盐和H+,其具体反应如下所示:
4NH4++6HCHO=(CH2)6N4H++3H++6H2O
生成的H+和(CH2)6NH4+:
Ka7.1X10-6(pH=5.6),可用NaOH标准溶液滴定。
(CH2)6NH4++OH-=(CH2)6N4+
+H2O
计量点时产物为六亚甲基四胺(CH2)6N4+,其水溶液显微碱性。
因此,在滴定终点时可采用酚酞作为指示剂,
达到终点时试液由无色变为微红色。
(2)
4.SO42-含量的测定
(1)重量法
将样品溶解于水后,用稀盐酸酸化后加热至沸,在不断搅拌下慢慢的加入氯化钡溶液,所得沉
淀经过过滤、洗涤、烘干、灼烧后,以硫酸钡形式称量,即可求得样品中硫酸根的含量。
Ba2++SO42-==BaSO4(白色)
(2)EDTA络合滴定
在酸性条件下,先用过量的氯化钡将溶液中SO42-的完全沉淀,溶液经过滤,沉淀硫酸钡经洗涤后,将其溶解于加入定量氨水的EDTA溶液中,过
量的EDTA以锌标准溶液回滴,以铬黑T为指示剂,可测定溶液中硫酸盐的浓度。
硫酸盐质量浓度(mg/L)=(V0-V1)C>
o<
96.06X1000/V式中:
V水样的体积,mL;
V0滴定空白
溶液所消耗锌标准溶液的体积,mL;
VI滴定样品所消耗锌标准溶液的体
积,mL;
C锌标准溶液的浓度,mol/L
、实验仪器和药品
(1)硫酸亚铁铵制备实验:
电子天平,量筒(10mL,5OmL),烧杯(50mL),锥形瓶(250mL),蒸发皿(60mL),玻璃棒,铁架台,酒精灯,大烧杯(可用水浴锅代替)、三脚架,石棉网,胶头滴管,药匙,吸滤瓶,比色管(25ml),吸量管,布氏漏斗,真空泵,温度计。
盐酸(2mol/L),硫酸(3mol/L),标准Fe3+溶液(0.0100mg/mL),硫氰酸钾(KSCN,1mol/L),硫酸铵(s),碳酸钠(10%),铁屑,乙醇(95%),pH试纸、滤纸。
(2)铵根离子含量测定:
25mL碱式滴定管;
250mL容量瓶;
25mL移液管;
1000mL试剂瓶;
250mL锥性瓶、烧杯;
10、100mL量筒;
洗瓶;
玻璃棒;
滴管;
表面皿;
吸耳球;
托盘天平;
电子分析天平。
氢氧化钠(AR);
邻苯二甲酸氢钾;
0.2%乙醇酚酞溶液;
40%甲醛;
硫酸铵、氯化铵试样,酚酞指示剂。
(3)SO42-离子含量的测定(重量法)
电子天平、烧杯、吸量管、表面皿、玻璃棒、胶头滴管、中(慢)速定量滤纸、玻璃漏斗、瓷坩
锅、干燥器、试管。
2mol/LHCI溶液、BaCb?
H2O122g/L、0.1mol/L硝酸银、O.025mol/LEDTA、甲基红指示剂(2g/L),乙醇。
(4)亚铁离子含量的测定
仪器:
分光光度计;
1cm比色皿
柠檬酸三钠水溶液,150g/L;
盐酸羟胺溶液,
50g/L;
盐酸溶液,3moI/L;
氨水溶液,2.5%;
1,10-邻菲啰啉溶液,2.5g/L:
称量2.5g1,10-邻菲啰啉溶于80C的约l00ml水中,加lml浓盐酸,冷却后加水稀释至1000ml,储于阴凉处备用;
醋酸-醋酸钠缓冲溶液:
称量272g醋酸钠
(NaCH3COZ3H2O)于约500m1水中,加入冰醋酸240ml,加水稀释至1000ml;
Fe2+标准溶液,lmg/ml:
称量7.024g硫酸亚铁铵于约500ml水中,加入浓盐酸10ml,移入l000ml容量瓶中,稀释至刻度;
Fe2+标准溶液,20?
g/ml:
吸取lmg/ml的亚铁标准溶液20ml于1000ml容量瓶中,用水稀释至刻度,混匀,临用前配制。
四、实验步骤
1.Fe屑的净化用台式天平称取2.0gFe屑,放入锥形瓶中,加入15mL10%W2CO3溶液,小火加热煮沸约10min以除去Fe屑上的油污,倾去Na2CO3碱液,用自来水冲洗后,再用去离子水把Fe屑冲洗干净。
2.FeSO4的制备往盛有Fe屑的锥形瓶中加入15mL3molL-1H2SO4,水浴加热至不再有气泡放出,趁热减压过滤(思考题4),用少量热水洗涤锥形瓶及漏斗上的残渣,抽干。
将滤液转移至洁净的蒸发皿中,将留在锥形瓶内和滤纸上的残渣收集在一起用滤纸片吸干后称重,由已作用的
Fe屑质量算出溶液中生成的FeSO4的量。
3.(NH4)2SO4FeSO46H2O的制备根据溶液中FeSO4的量,按反应方程式计算并称取所需(NH4)2SO4固体的质量,加入上述制得的FeSO4溶液中。
水浴加热,搅拌使(NH4)2SO4全部溶解,并用3molL-1H2SO4溶液调节至pH为1〜2(思考题5),继续在水浴上蒸发、浓缩至表面出现结晶
薄膜为止(蒸发过程不宜搅动溶液)。
静置,使之缓慢冷却(思考题6),(NH4)2SO4FeSO46H2O晶体析出,减压过滤除去母液,并用少量95%乙醇洗涤晶体(思考题7),抽干。
将晶体取出,摊在两
张吸水纸之间,轻压吸干。
观察晶体的颜色和形状。
称重,计算产率。
4.产品检验]Fe(叫的限量分析]
⑴Fe(m)标准溶液的配制。
称取0.8634g
NH4Fe(SO4)212出0,溶于少量水中,力口2.5mL浓H2SO4,移入1000mL容量瓶中,用水稀释至刻度。
此溶液为0.1000gL-1Fe3+。
(2)标准色阶的配制。
取0.50mLFe(m)标准溶液于25mL比色管中,力口2mL3molLHCl和ImL25%的KSCN溶液,用蒸馏水稀释至刻度,摇匀,配制成Fe标准液(含Fe3+为0.05mgg-1)。
同样,分别取0.05mLFe(川)和2.00mLFe(川)标准溶液,配制成Fe标准液(含Fe3+,分别为
l1
0.10mgg-、0.20mgg-)。
(3)产品级别的确定。
称取1.0g产品于25mL比色管中,用15mL去离子水溶解,再加入2mL3molLHCl和lmL25%KSCN溶液,加水稀释至25mL,摇匀。
与标准色阶进行目视比色,确定产
品级别。
此产品分析方法是将成品配制成溶液与各标准溶液进行比色,以确定杂质含量范围。
如果成品溶液的颜色不深于标准溶液,则认为杂质含量低于
某一规定限度,所以这种分析方法称为限量分析。
5.(NH4)2SO4FeSO46H2O含量的测定
⑴(NH4)2SO4FeSO46H2O的干燥。
将步骤3中所制得的晶体在100C左右干燥2〜3h,脱去结晶水。
冷却至室温后,将晶体装在干燥的称量瓶中。
(2)&
Cr2O7标准溶液的配制。
在分析天平上用
差减法准确称取约1.2g(准确至0.1mg)K2Cr2O7,放入100mL烧杯中,加少量蒸馏水溶解,定量转移至250mL容量瓶中,用蒸馏水稀释至刻度,计算K262O7的准确浓度。
c(K2CDO7)
(NH4)2SO4FeSO46H2O两份,分别放入250mL锥形瓶中,各加100mLH2O及20mL3molLH2SO4,
加5mL85%H3PO4,滴加6〜8滴二苯胺磺酸钠指示剂,用K2Cr2O7标准溶液滴定至溶液由深绿色变为紫色或蓝紫色即为终点。
6c(K2Cr2O7)V(K2Cr2O7)M(Fe)
m(样)
w(Fe)=
6•铵根离子含量测定
(1)试剂的配制
酚酞指示剂:
称取0.58酚酞溶于50mL95%乙醇和50mL的水溶液中。
0.1mol/LNaOH溶液:
在台称上取NaOH固体2g于小烧杯中,加入50mL蒸馏水使其溶解,稍冷后转入500mL试剂瓶中,加水450mL,用橡皮塞塞好瓶口,摇匀。
NaOH溶液的标定:
准确称取0.4~0.5g邻苯二甲酸氢钾三份(分析天平;
差量法;
有效数字,小数点后四位),分别置于250mL锥形瓶中,加水40~50mL溶解后,滴加酚酞指示剂1~2d,用NaOH溶液滴定至溶液呈微红色,30秒内不褪色,即为终点。
平行测定三份。
NH4CI和(NH4)2SO4混合试样:
用电子天平准确地称量0.15-0.20gNH4CI和0.85-0.90g
(NH4)2SO4固体于小烧杯中。
用大约30ml蒸馏水将固体溶解定容于250.00mI容量瓶中。
(2)实验步骤
用移液管移取25.00mL溶液于250mL锥形瓶中,加入5ml中性甲醛溶液,1〜2滴酚酞,摇匀,静置1min后,用0.10mol/LNaOH标准
溶液滴定至溶液呈淡红色且半分钟不褪色为终点。
平行测定三份,计算试样中的含氮量及相对平均偏差。
(3)数据处理
(1)NaOH的标定
序号、
数据
项目
1
2
3
mKHp/g
0.3198
0.4492
0.4444
VNaOH/ml
16.65
23.28
23.15
CNaOH/mol/L
0.09415
0.09458
0.09410
0.09427
「di
0.00012
0.00031
0.00017
相对平均偏
差/%
0.21
V(试样)/ml
25.00
V(NaOH)/ml
12.88
12.84
12.92
C(NH4+)
0.06476
0.06456
0.06496
C(NH4+)/mol/L
1di
0.00000
0.00020
相对平均偏差
/%
7.SO42离子含量的测定
7.1试剂的配制
硝酸银溶液C(AgN03)=O.lmol/L:
称取1.7160g硝酸银,溶于蒸馏水中,在容量瓶中稀释至100mL,贮于棕色瓶中。
甲基红指示剂(2g/L):
0.2g指示剂溶于1O0ml6O%乙醇中。
0.025mol/LEDTA:
称取4.0g乙二胺四乙酸二钠于500ml烧杯中,加200ml水,温热使其溶解完全,转移到500ml容量瓶中用蒸馏水定容。
7.2实验步骤
(1)空瓷坩锅的恒重:
洗净两个瓷坩锅,晾干,
编号,然后在800〜850C马沸炉中灼烧。
第一次灼烧时间30〜45min,取出放冷后,转入干燥器中冷却至室温后称重,然后再放入同样温度的马沸炉中,进行第二次灼烧时间15〜20min,取出放冷后,转入干燥器中冷却至室温后再称重。
如此操作直到相邻两次称量结果相差不超过0.3mg,即
为恒重。
(2)沉淀的制备:
称取50g氯化钡样品,精确0.1g,置于400ml烧杯中,加200ml水溶解,加2滴甲基红指示剂,滴加盐酸溶液至刚变红,再过量2ml,
在石棉网上加热近沸,勿使溶液沸腾,以防溅失,冷却,用中速滤纸过滤,用水洗涤5次,每次约
10ml,将滤液和洗液收集到500ml烧杯中,加热至近沸,在不断搅拌下,先加入6mlEDTA溶液,再慢慢加入25ml试样,继续沸腾15min,冷却,盖上表面皿,将玻璃棒靠在烧杯嘴边,沸水浴上保温2h(要经常不断搅拌)。
(3)溶液自然冷却后,室温下用慢速定量滤纸过滤,先将上层清液倾注在滤纸上,再用温水洗涤3~4次,每次用水约5~10ml,洗涤时均用倾泻法过滤。
然后将沉淀小心转移至滤纸上,并用一小片滤纸擦净烧杯壁(该滤纸是折叠滤纸时撕下的小片)。
将此滤纸片放入漏斗内的沉淀上,再用水洗涤沉淀至无氯离子为止(检查方法:
用小试管收集2ml滤液,加入2滴硝酸银,若无白色沉淀产生,表示氯离子以洗净)。
(4)利用玻璃棒把滤纸和沉淀从漏斗中取出,折卷成小包,把沉淀包在里面,特别注意,勿使沉淀有任何损失。
将滤纸包装进已质量恒重的坩埚中。
使滤纸层较多一边向上。
在电炉上干燥、炭化,在8O0~850C马沸炉中灼烧至恒重(方法同空坩埚的恒重)。
3.4数据处理
SO42-离子含量的测定
试样质量/g
(0.1787+0.8513)/10=0.1030
灼烧后坩埚质量/g
30.9452
沉淀和坩埚总量/g
31.0508
沉淀质量/g
0.1056
SO42—质量分数/%
60.30
C(SO42—)/mol/L
0.02588
8.亚铁离子含量测定
(1)吸光光度法
(一)工作曲线的绘制量取20?
g/ml的亚铁标准溶液0.00m1、2.50m1、5.00ml、10.00ml、20.00ml(相当于分别含0、50、100、200、400?
g/Fe2+)分别加入l00ml烧杯中,用水稀释至50ml,加入150g/L柠檬酸三钠溶液5m1,用3mol/L盐酸或2.5%氨水溶液调节溶液pH为2.4~2.6,加入50g/L盐酸羟胺溶液5ml混匀,加入1,10-邻菲罗琳溶液5m1,加入醋酸-醋酸钠缓冲溶液l0ml,将溶液移入到l00ml容量瓶中,用水稀释至刻度,混匀放置60min。
用分光光度计在波长506nm处用lcm比色皿,以水为参比溶液测定该标准系列的吸光度,以Fe2+
标准溶液浓度(?
g/100ml)为横坐标,以其对应吸光度作纵坐标绘制工作曲线。
(二)湿法磷酸中铁含量的测定
吸取1ml湿法磷酸,用水稀释至100m1,混匀,移取1m1到100m1的烧杯中,用水稀释至50m1,以下操作同工作曲线的绘制,测定其吸光度。
不加试样,在同样条件下进行空白试验。
(三)计算总铁含量按下式计算
(叫-那単』
式中:
m1为从工作曲线上查得被测试液Fe的质量,?
g;
m0为从工作曲线上查得试剂空白溶液中Fe的质量,?
m为吸取试样溶液相当于试样的质量,g
(2)重铬酸钾法
1.配制K2Cr2O7标准溶液
准确称取固体0.9〜1.1g置于250ml烧杯,加50ml水溶解,定容200ml容量瓶中,摇匀。
(思考题1)
2.亚铁盐中铁含量的测定
(1)准确称取0.8~1.2g试样3份于锥形瓶中,加50ml水,10ml硫酸,5ml磷酸,加6d二苯胺磺酸钠;
(2)用K2Cr2O7标准溶液滴定至溶液由绿色突变为紫色或紫蓝色为终点;
)
(3)计算浓度。
五、注意事项
1.不必将所有铁屑溶解完,实验时溶解大部分铁屑即可。
2.酸溶时要注意分次补充少量水,以防止FeSO4析出。
3.注意计算(NH4)2SO4的用量。
4.硫酸亚铁铵的制备:
加入硫酸铵后,应搅拌使
其溶解后再往下进行。
加热在水浴上,防止失去结晶水。
5.蒸发浓缩初期要不停搅拌,但要注意观察晶膜,一旦发现晶膜出现即停止搅拌。
6.最后一次抽滤时,注意将滤饼压实,不能用蒸馏水或母液洗晶体。
 升级会员
升级会员 